
slurm 作业数组的正确使用姿势
最近一次想要批量运行任务前试着运行了一个个例,发现占用的内存意外的比较多,所以一口气把所有提交到服务器上显然不太妥当。由于之前在服务器上用 slurm 仅限于 srun 而从来没有尝试过 sbatch,所以就对这方面的知识进行了一番恶补,正好也能补充十二月份的博客内容。
如何确定内存是否充足对于当前节点,使用 top 和 htop 就能得到相关的信息,但对于其他节点,如果没有登录权限,那么这种实时监控的手段就没法派上用场了。
这时候,可以使用 free -h 命令将节点在该时间点的内存使用情况信息打印出来:
1234$ srun -w nodexx free -h total used free shared buff/cache availableMem: 487G 144G 2.7G 162M 340G 340GSwap: 49G 685M 49G
重点关注 availa ...

DNA 语言模型 GPN 在突变效应预测中的应用及表现
众所周知,语言模型已经在诸多领域得到了广泛应用。受深度学习狂潮影响深远的生物学领域里,出色的语言模型应用不断涌出。以 DeepMind 为例,其所开发的 Alphafold、Alphamissense 等都是基于语言模型实现的。
这篇文章将分享一个最近发表的 DNA 语言模型(GPN,Genomic Pre-trained Network)和它的变体 GPN-MSA,该模型与之前的 DNABERT 等 DNA 语言模型不同,其使用单个碱基作为 token 进行训练,并在无监督的突变效应预测中取得了良好的成效。
相关论文链接见文章结尾,本文章将重点介绍两个模型的设计和它们之间的区别,并写出一些很值得学习的地方。
GPNGPN 是一种自监督训练的 DNA 语言模型,与 GPN-MSA 不同的地方在于,它可以仅依靠未经过比对的数个基因组序列而不使用多序列比对(MSA)进行训练,此外它使用的为单纯的 DNA 序列而未使用到任何功能基因组信息,因此可以说 GPN 的泛用性是非常强大的,因为对于很多类群中的生物来说,获得跨物种的 MSA 和全面的功能基因组数据并不是容易的事情。
GPN 的模型设计 ...
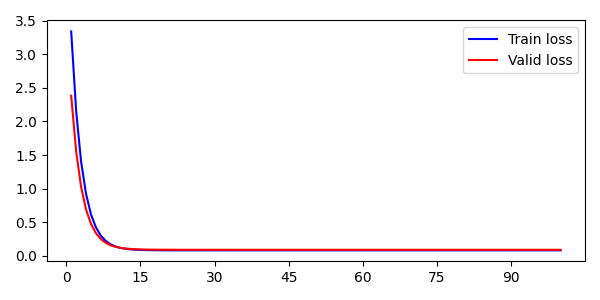
深度学习 loss 追踪器(用于追踪 loss 变化)
最近设计了一个深度神经网络,虽然使用 logging 模块记录 Train loss 和 Valid loss 也算方便,但作图观察 loss 的变动趋势还是要更加直观的,考虑到网上貌似没有这方面的方法,这里放一下自己的记录方式。
1234567891011121314151617181920212223242526272829303132import matplotlib.pyplot as pltfrom matplotlib.ticker import MaxNLocatorimport timeclass loss_tracer(): def __init__(self, outpath): self.train_loss_list = [] self.test_loss_list = [] self.outpath = outpath timestamp = time.time() self.starttime = time.strftime('%Y_%m_%d_%H_%M', time.localtime(timestamp)) def __call__(sel ...

Python 循环太费时?让多进程解决这个问题(concurrent)
前言这篇文章主要分享如何使用 Python 的 concurrent 库进行多进程运行以加速分析。在约一年前我在跑多个物种的 ABBA-BABA test 时遇到过分析缓慢的问题,因为涉及到的物种有九十多种,所以在经过筛选以后四物种组合的数量依然高达数百万,但由于那个分析并不是很要紧所以也就没有多管(最后跑了一个多月)。
现在的状况与那时有些许不同,最近跑的分析大多涉及到大量计算,由于比较急迫地想看到最终结果,我不得不学习多进程方法来进行加速,很多时候进步都需要一些些小小的契机,now it comes。
由于 Python 并不是从底层框架开始让人写代码,所以它在拥有友好入门门槛的同时也牺牲了一定的性能,特别是在处理计算密集型任务时,Python 比起 C 来说就显得有些捉襟见肘。
当然我肯定是不愿意写 C 的代码的,一是因为我只学了一些皮毛,二是因为相比之下 Python 实在太方便,俗话说性能不够进程来凑,既然我跑的不够快,那我就多跑一些,以量胜质。
在使用多进程前,建议先检查一下自己的代码是否可以进行优化,有时候换成合适的数据操作方式就可以让分析的速度有质的飞跃,使用多进程应 ...

Python seaborn 绘制复杂多变量图(hexbin + 拟合直线)
当初作图时想往 sns.pairplot 里加些内容,搜索了解到 sns.PairGrid 可以更灵活地绘制多因素(多变量)关系图,遂在网上找相关教程,最后发现 CSDN 上的内容竟然都要付费,无奈转向 ChatGPT 求助,最后在 C 老师帮助下成功完成了自己的想法。
以上既是这篇文章的来源也是动机,这篇文章就来分享一下如何使用 sns.PairGrid 绘制更加复杂的多变量图,本文示例将是 hexbin + 拟合直线,但只要知道如何将图架构起来那么绘制其他类型的图也不会有问题。
sns.pairplot 的详尽用法:
https://seaborn.pydata.org/generated/seaborn.pairplot.html#seaborn-pairplot
一般的多变量图绘制:
12345678910111213141516171819import numpy as npimport pandas as pdimport matplotlib.pyplot as pltimport seaborn as sns# 模拟三个变量np.random.seed(114514)a ...

关于人类基因组注释中起始和终止位点相同的现象
前言已经有一个月没有更新文章,之前在知乎看到过一段话,大体是说写博客最重要的不是怎么把博客做的好看,而是如何持之以恒地产出,这句话让我感触颇深,正值国庆期间放假,就记录一下这段时间在分析数据时候发现的东西,以后万一有其他朋友发现并疑惑也好快速找到答案。
这里进行一个简要的概括:
长度为 1 并不代表该构成该基因的所有片段组合长度为 1,例如 1bp 的 exon 可能是微外显子,在剪切后与其他 exon 组合共同构成成熟 mRNA。
CDS 长度不为 3 的整数倍,说明该基因的 CDS 注释不完善,例如缺失 3' 或 5' 端。
出于第 2 点现象,建议在下载注释时,选择 basic 版本,因为该版本中所有的注释都是完善的。
基因组注释中长度为 1 的现象解释在使用基因组注释(版本:https://www.gencodegenes.org/human/release_19.html )时,我发现了两个问题:
有些 CDS 的长度为 1。
CDS 的长度不一定为 3 的倍数。
由于我是先发现的第一个问题,此后才发现的第二个问题,因此顺序如上,首先看一看第一个问题:
12345 ...

JuseKit(八) —— 计算转录组组装指标
generatePortalLinks(8);
更新变动及进度JuseKit 故事传之开学第一天,我在课题组实验室搞新功能。
已有功能的相关教程请见:Juseの软件开发
本文所涉及功能借鉴了 Trinity 脚本的输出格式。
本次更新变动
完善了部分报错提示。
完善了部分文本显示。
新增了转录组组装指标计算功能。
目前的功能进度
提取最长转录本。
根据 id 提取序列。
对序列的 id 进行各种处理。
串联序列并得到分区信息。
批量进行序列格式转换。
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。
批量进行序列的物种数和长度过滤。
火山图绘制。
气泡图绘制。
组装指标计算。
叠盾警告?:本软件解释权归属 Juse 所有,本软件能走多远具体得看 Juse 能坚持多久。
下载地址:https://github.com/JuseTiZ/JuseKit/releases
转录组组装指标计算本文主要着重于这次更新新增的功能,其他模块请走这里。
要求的数据需要输入的数据为 fasta 格式的序列文件。
可通过将文件拖拽至文本框或者点击右侧按钮读取文 ...

基于 js 在系列文章开头设置文章传送门(hexo + butterfly)
示例效果
generatePortalLinks(3);
注:以上超链接并没有任何指向,只作示例用!
前言事实上这个想法在很早以前就有了,但那时候我还不知道具体的实行方案(只知道最笨的方法是直接在每篇文章开头自己写传送门,不过每次更新一篇新的文章以后就要全部都补充一遍,非常麻烦)。在经过博客一系列的魔改以后,也算是储备了一小些基本的 css js 知识,所以就有了这篇文章。
js 实现首先 hexo-theme-butterfly\source\js(请替换成自己的 js 文件存放路径)中新建一个 portallinks.js 文件,在里面填写以下内容:
12345678910111213141516171819202122232425262728293031function generatePortalLinks(currentArticleId) { var links = [ //填写 id 及对应的标题和链接 { id: 1, title: "这里填写标题 1", url: "这里填写链接 1" }, { id: 2, t ...
JuseKit(七) —— 绘制 GO 富集分析气泡图
generatePortalLinks(7);
更新变动及进度和上次更新火山图功能正好相差一个月,也算是勉强符合一开始二至四周一更的预期?
这次更新说实话还是遇到了很多困难的,最直观的体验应该是用 R 画气泡图可比用 Python 方便多了。
不过既然已经引入了绘图的库,那么就理应尽善尽美,所以这一次的更新正式上线 —— 绘制富集分析气泡图。
已有功能的相关教程请见:Juseの软件开发
使用 R 绘制气泡图的教程可见:比较转录组分析(六)—— GO 富集分析与可视化
本次更新变动
修复了 ‘火山图绘制’ 模块中文件不能通过点击按钮读取的问题。
新增了气泡图绘制功能。
新增了 GO id 注释功能(23.8.27)。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(已实现)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(已实现)
批量改后缀。(已实现)
批量进行序列格式转换。(已实现)
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。(已实现)
批量进行序列的物种数和长度过滤。(已实现)
火山图绘制。 ...
DiscoVista 可视化系统发育不一致
关于系统发育不一致基因树不一致是一个常见的问题,导致这个问题的原因有很多,如上一篇文章中提到过的谱系不完全分选和渐渗,此外也可能是因为:
水平基因转移(Horizontal Gene Transfer)。
基因遗传方式的差异(核基因和线粒体基因)。
有时我们会想要找出物种树和基因树间的不一致在哪以及有多严重,因此我们需要一个可以可视化并且能够提供良好解释性的工具。
所以这篇文章将介绍 DiscoVista(Discordance Visualization Tool),它可以帮助我们可视化特定类群在不同树间的拓扑不一致情况。
DiscoVista 的安装和使用DiscoVista github:https://github.com/esayyari/DiscoVista/tree/master
详细的示例教程:https://github.com/esayyari/DiscoVista/tree/master/example
前排提示,该文所使用的 DiscoVista 经过了博主本人的一些修改以方便使用,如果想要使用和作者一样的版本请更改安装步骤为:
1234$ git clo ...
使用 IQTREE 计算一致性因子
关于谱系不完全分选和渐渗
注意:该前言部分使用 ChatGPT4.0 生成并由 Juse 纠正总结,图源自维基百科。
问题:请介绍一下谱系不完全分选(ILS)和渐渗(introgression)对推断系统发育关系的影响,随之引出基因一致性因子(gene concordance factor)和位点一致性因子(site concordance factor)的概念并介绍它们的作用。
在生物学中,推断不同物种的系统发育关系是一个核心课题。然而,系统发育树的构建很复杂,可能受到多种因素的影响。这里我们着重介绍两个重要因素:谱系不完全分选(Incomplete Lineage Sorting,简称 ILS)和渐渗(Introgression)。
谱系不完全分选 (ILS): 谱系不完全分选发生在物种分化的过程中。当两个物种在很短的时间内分化时,一些基因座可能没有足够的时间来完全分化,因此这些基因座在不同物种之间可能会保留共同的祖先多态性。这意味着即使两个物种已经分化,它们的一些基因序列可能仍然相似。这在系统发育树的构建中会引入混乱,因为它可能导致基于这些基因的树与真实的物种树不一致。
...
贝叶斯建树之 Mrbayes 篇
贝叶斯建树原理及优缺点
注意:该前言部分使用 ChatGPT4.0 生成并由 Juse 纠正总结。
问题:请介绍一下系统发育分析中的贝叶斯推断法(原理、优缺点等),并指出几个代表生信软件。
在系统发育学中,贝叶斯推断是一种常用的统计方法,用于根据观测数据推断出物种间的系统发育关系。
原理:
贝叶斯推断的原理基于贝叶斯定理,该定理可以将先验概率和似然度结合起来,得到后验概率。
在系统发育分析中,贝叶斯推断法被用来估计种群或物种之间的演化关系。这种方法假设所有可能的树形图都存在某种先验概率,并使用蒙特卡洛马尔科夫链(MCMC)来在所有可能的树形图和其关联参数的空间中进行采样。每一步,系统会根据特定的概率提议一种新的树形图或参数,然后根据新的树形图或参数与当前状态的拟合度决定是否接受。
优点:
贝叶斯推断提供了一种直观的方式来理解不确定性和证据的权重。因为后验概率可以直接解释为在给定数据下模型或参数的概率。
贝叶斯推断允许使用先验信息,有助于引导分析并提高准确性。
缺点:
计算复杂性高,尤其是对于大数据集。MCMC方法需要长时间的运行才能确保采样的充分性。
对先验概率的选择可能对结 ...
JuseKit(六) —— 绘制火山图
generatePortalLinks(6);
更新变动及进度很久没更 JuseKit,这一段时间我也时不时在想我还能搞什么出来,或者说我最应该搞什么。很多其他有用的功能事实上在别的软件诸如 BTtools Phylosuite 中都有涵盖到,所以陷入了一段瓶颈期(说到底是不够勤勉)。
恰巧毕业以后已经没啥事,闲余之时就思考这些事情,想着既然已经把这个坑挖开来了,那就也不要想太多直接把最开始的设想全部完成一遍再说吧。
所以这一次的更新正式上线 —— 绘制火山图。
已有功能的相关教程请见:Juseの软件开发
本次更新变动
修复了 ‘序列过滤’ 模块中文件不能拖拽读取的问题。
新添了日志模块,报错后将有 error.log 出现在程序根目录。
新增了画图模块,并完成了火山图绘制的功能。
新增的模块导致软件大小的膨胀,对于不需要绘图且内存紧张的人来说可能不是好消息。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(已实现)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(已实现)
批量改后缀。(已实现)
批量进行序列格式转换。(已实现)
批 ...
Astral 建树指南
系统发育分析各类方法系统发育关系重建的方法有很多,例如最大似然法(ML)、贝叶斯推断法(BI)、邻接法(NJ)、最大简约法(MP)等,每种建树方法各有其特定优缺点,在很多文章中也能看到作者会采用多种不同的方法进行物种树重建以作为一种交叉验证方法。本文着重介绍其中一个经常出现的软件 Astral。
Astral 是一种 Coalescent-based method,与之不同的另一种方法为 Concatenated-based method:
Coalescent-based method: Coalescent-based 方法是一种模拟基因座的共同祖先的过程来构建进化树的方法。在这种方法中,每个基因座可能有自己独立的进化历史,而不是假设整个基因组有一个统一的历史,最后得到的物种树是这些基因座的统计整合。这种方法通常用于处理物种树和基因树的不一致性,比如基因水平转移、混合和不完全谱系分选。这是一种比较复杂的方法,通常在研究种群遗传学和进化过程时使用。
Concatenated-based method: Concatenated-based 方法是将多个基因或基因片段首尾连接起来 ...