
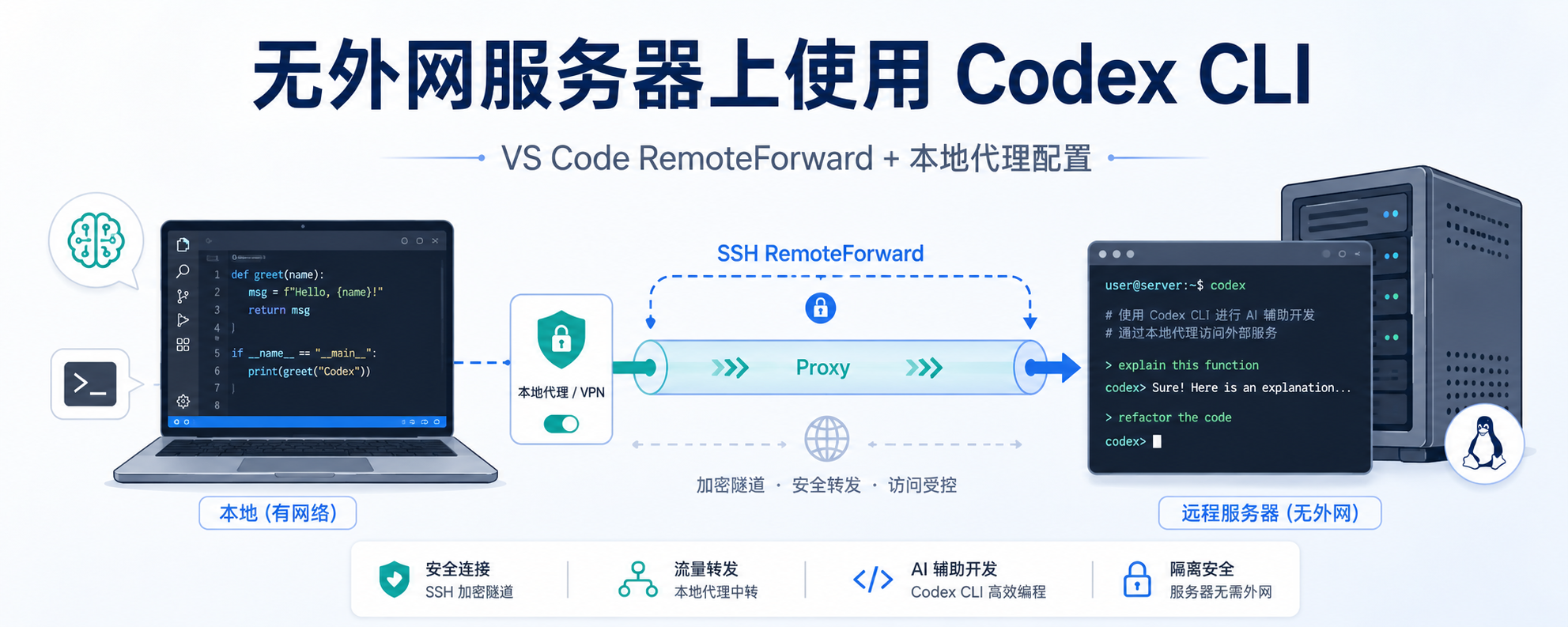
远程服务器无法连外网?用 VS Code RemoteForward 跑通 Codex CLI
整活开头:你是否还在为课题组服务器上无法连外网而使用不了 GPT + Codex cli 而发愁?你是否还在因自己冲了 GPT plus 而无法充分利用 Codex 额度而锤头顿足?跟着本文,我将手把手教你如何在无法连外网的远程服务器上使用 GPT + Codex cli 进行 vibe coding!
整活结束,其实这类教程在网上也已经有一些资源了,但是多多少少有些细节没讲清楚 or 有很多后续容易出问题的点没有讲清楚,所以干脆自己做个更详细的教程出来,也算水一篇博客。
前提需求:
订阅了 GPT plus 及以上的套餐。
有一个自己的 VPN/机场/梯子。
使用的 IDE 为 VScode(本文的示例是以 VScode 为准的)。
流程1. 安装 Codex cli安装 Codex cli 并不困难,可以参考我在前不久发的文章进行操作,仅需把命令换为 npm install -g @openai/codex。
2. Remote config 中配置 RemoteForward通过配置 RemoteForward 可以让远端服务器走本地主机的某个端口,这是最关键的一步。首先点击 ...
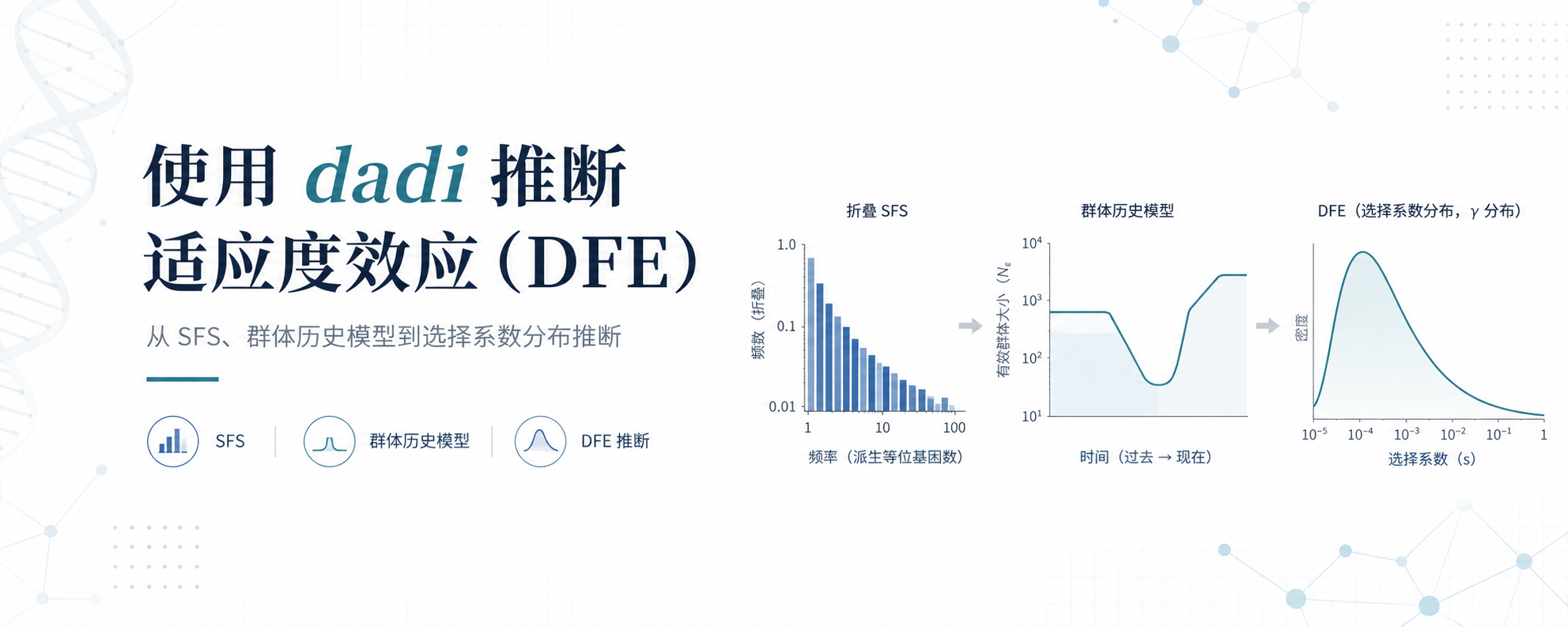
使用 dadi 推断适应度效应(DFE)
前言适应度效应分布(Distribution of Fitness Effects,DFE)是群体遗传学中的一个核心概念,用于描述新产生突变对生物适应度的影响大小及其分布情况。一般认为,大多数新突变是近中性或有害的,只有极少数能够带来适应性优势。准确推断 DFE,不仅有助于理解自然选择如何塑造遗传多样性,也为遗传负荷评估、疾病变异解释以及进化历史研究提供了重要依据。
在这类分析中,许多基于位点频谱(Site Frequency Spectrum,SFS)的 DFE 推断方法得到了广泛应用。这类方法通常利用中性位点与受选择位点的频谱差异,通过建立群体遗传学模型来估计选择系数的分布。在一个典型的 DFE 推断流程中,通常需要:
构建中性位点和目标位点的位点频谱(SFS);
根据中性位点估计群体历史(demographic history)参数;
在固定人口历史模型的基础上,拟合受选择位点的选择系数分布;
根据模型参数推断不同强度有害突变所占的比例,并进一步开展生物学解释。
本文将介绍一个常被用于 DFE 推断的群体遗传学软件 —— dadi。
dadi 使用文章:Inferring t ...
Claude code 在远程服务器上的安装(无root)
写这篇文章的原因是,我按照网上一些已有教程操作在服务器上没法成功安装 Claude code,问题出在自己下载 Nodejs 的包并调用 npm 时会出现以下报错:
1/usr/bin/env: node: No such file or directory
亦或者是:
123node: /lib64/libm.so.6: version `GLIBC_2.27' not found (required by node)node: /lib64/libc.so.6: version `GLIBC_2.28' not found (required by node)node: /lib64/libc.so.6: version `GLIBC_2.25' not found (required by node)
究其原因可能是我的一些环境配置过于紊乱导致出现冲突,尝试了一些其他的方法发现有更简单的安装方式,固有此文。
安装方案整体的安装方案其实很简单,首先用 mamba/conda 安装一个只有 nodejs 的纯净环境:
12conda create - ...

hexo+butterfly 博客评论系统迁移(Valine+LeanCloud 到 Waline+Neon)
前言在前不久,LeanCloud 发布公告,宣布其停止新用户注册和新应用创建,且将于 2027 年 1 月 12 日停止服务。
此时我刚捣鼓好 Valine 的邮件通知系统没有多久,但可惜的是我不得不面对现实。由于 Valine 官方基本沉寂,我大概率等不来一个好的替代方案(Valine 仅支持 LeanCloud 作为数据库)。在这段时间忙完自己的论文后,终于有了时间来处理博客事务,研究了一天成功将博客的评论系统进行了迁移,故有此文。
评论系统迁移一些可能需要提前注意的事项迁移到 Waline 可能需要你有一个自己的域名(因为要绑定一个子域名作为评论管理系统页面),如果你没有自己的域名,可以考虑购买一个或使用其他评论系统作为替代(即本文内容对你可能无用)。此外,本博客基于 hexo+butterfly 搭建,一些后续的调整可能在其他主题中不适用。
Waline 部署这一部分内容主要来自:Waline 官方教程 —— 快速上手,并针对部分内容进行细节补充。
部署服务端及创建数据库这两个部分基本与上述官方教程无异,虽然当前 Vercel 页面布局可能存在些许不同,但总体流程没有变化,所以 ...

使用 PheTK 进行全表型组关联分析
前言全表型组关联分析(PheWAS,Phenome-wide Association Studies)是一类用于系统性探索遗传变异与多种表型之间关联的研究方法。与常见的全基因组关联分析(GWAS)相比,二者的视角恰好相反:
GWAS 关注的是“针对一个表型,在基因组范围内寻找哪些变异与之相关”;
PheWAS 则关注“针对某个特定变异(或一类变异),在众多表型中寻找它会影响哪些性状”。
近年来,随着测序规模的扩大与统计方法的发展,稀有变异(rare variants)在复杂性状中的作用逐渐受到重视。由于单个稀有变异往往难以单独检出显著关联,研究者通常采用 burden analysis 等策略将同一基因中的有害变异聚合起来,以提升统计功效。在典型的 burden 分析流程中,通常会:
识别基因中的功能有害变异,例如预测的 loss-of-function 变异或机器学习模型给出的高致病性错义变异;
根据样本是否携带这些有害变异进行编码,编码方式可以是简单的二元(0/1),也可以使用携带数量作为计数值;
对编码后的变量在多种表型上执行关联分析,从而识别哪些表型与该类有害变异显著相关 ...
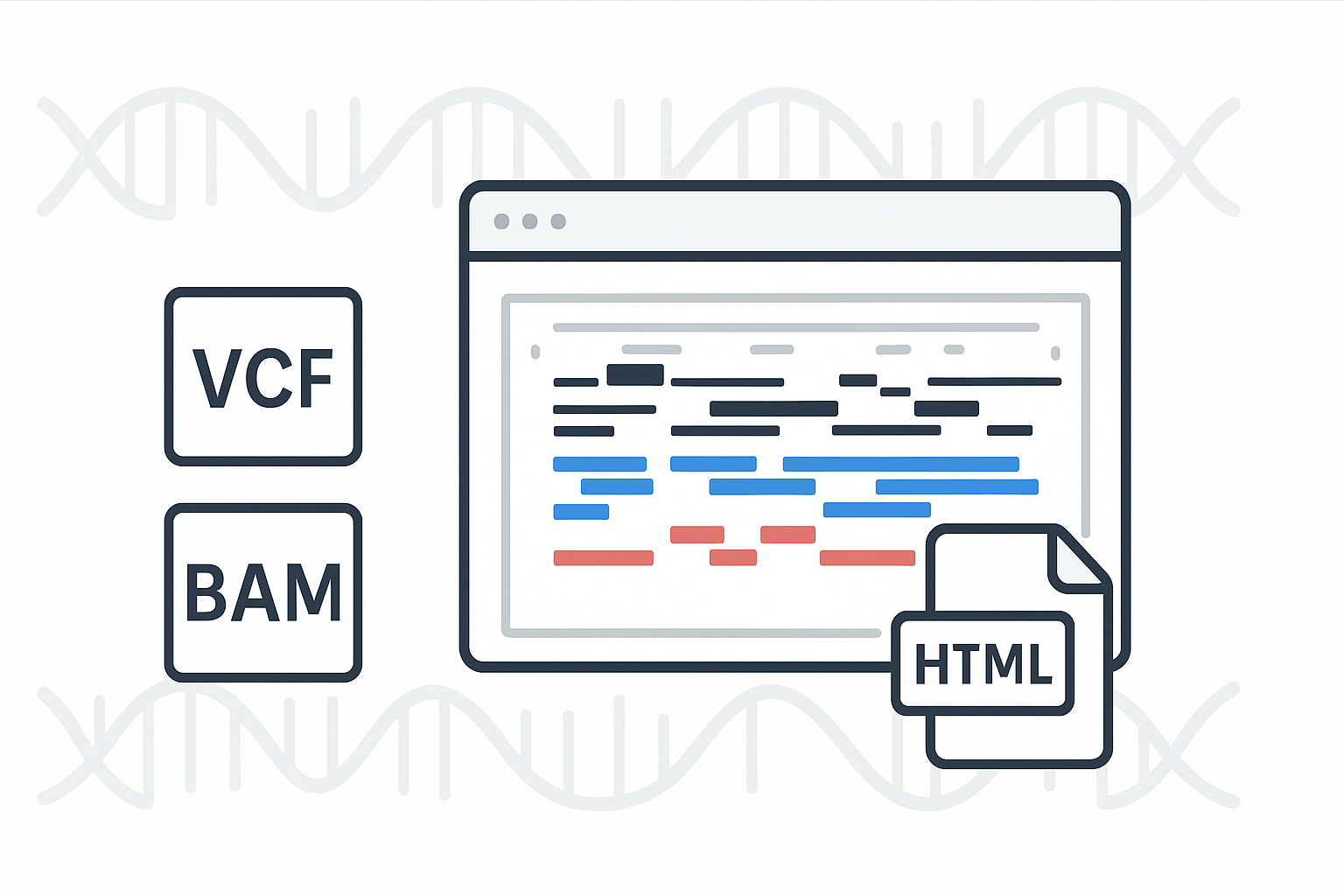
使用 igv-reports 进行基因组数据可视化
基因组数据分析中,Integrative Genomics Viewer(IGV)以其直观的交互式界面和丰富的可视化功能,成为我们查看变异和注释等基因组信息的首选工具。然而,在某些情景下 IGV 的可视化优势难以发挥:基因组数据在服务器上,但在服务器中无法方便地调用图形页面,而基因组数据下载到本地再使用 IGV 可视化又过于不便且耗时(特别是在数据量庞大的情况下)。
针对这一问题,igv-reports 成为了一个很好的替代工具。它能够将 IGV 的会话内容和基因组浏览快照一并打包成静态 HTML 报告。生成的报告文件下载到本地后,用户可直接在浏览器中查看和交互,操作体验与 IGV 类似,同时支持灵活分享,兼顾效率与便捷性。
Source: https://igvteam.github.io/igv-reports/examples/example_vcf.html
需要注意的是,igv-reports 适用于可视化基因组部分区域时使用,例如你关注的是某些变异位置或特定基因附近的比对情况/信号时,使用 igv-reports 是合适的。但如果你想要大尺度地进行可视化(例如展示整条 ...
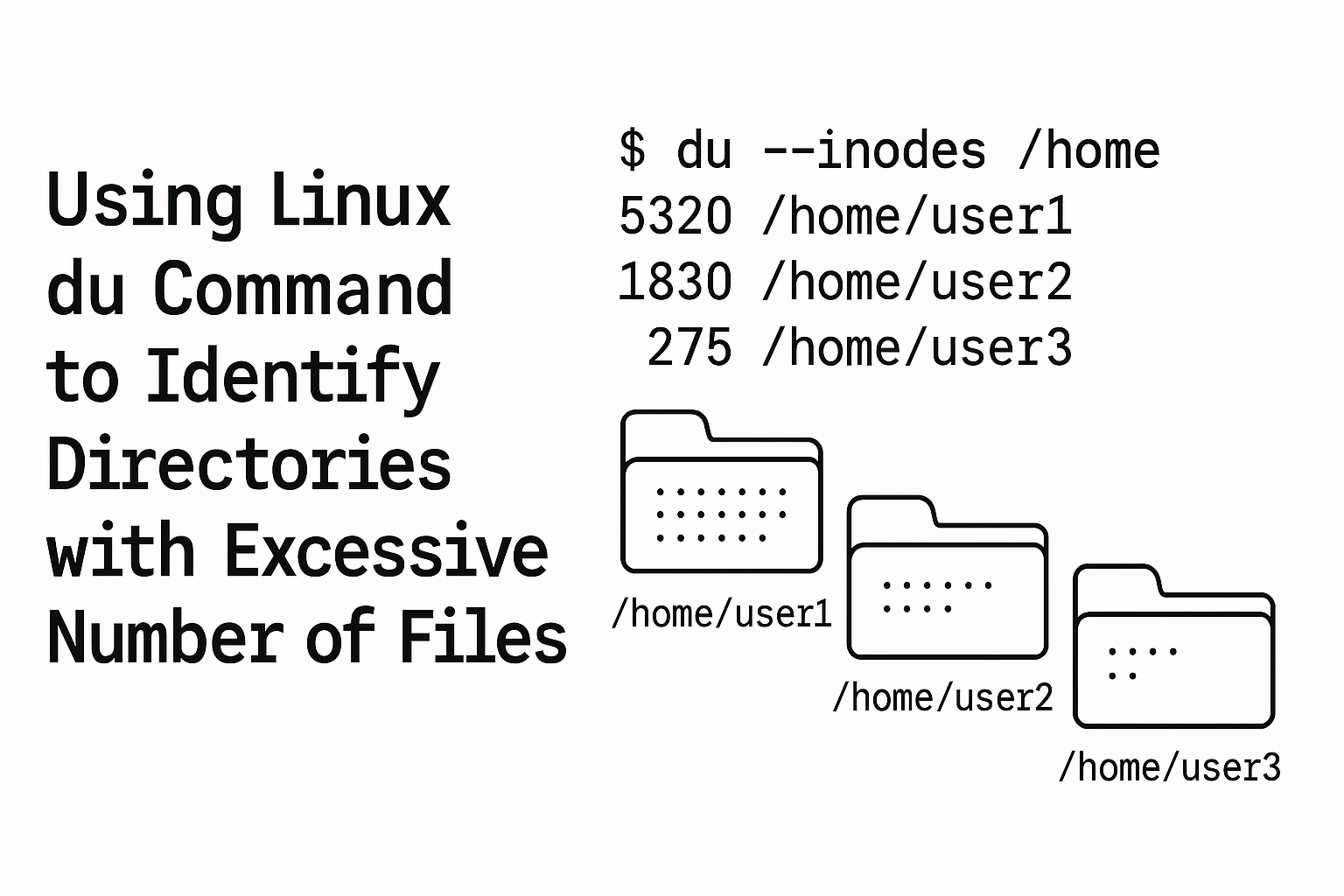
linux 中使用 du 排查文件数量过大的目录
这一段时间对服务器进行备份时发现有些目录的扫描速度非常慢,经调查这些目录大多都是某些 pipeline 在运行时产生的中间文件,虽然其存储占用并不大但由于文件小且多导致在处理这一部分目录时不必要地产生了过大的时间开销,遂在网上寻找定位这些目录的方法并觉得可以水篇博客,固有此文。
这篇文章将介绍如何使用 du 识别出那些包含文件数量过大的目录。
基础介绍每个文件/目录在 Linux 中都有一个 Inode(Index Node),文件系统创建时有固定的 Inode 总数,用尽会导致无法新建文件,而过大的 Inode 数量也会导致一些困扰(在我的应用场景中即部分命令的运行时间开销大幅增加),可以使用 df -ih 查看当前各个文件系统的的 Inode 情况:
123456$ df -ihFilesystem Inodes IUsed IFree IUse% Mounted ondevtmpfs 63M 1.1K 63M 1% /devtmpfs 63M 30K 63M 1% /d ...

trim_galore 后 Per Base Sequence Content 出现问题的原因
最近在处理基因组数据时,遇到的大多数据都已经处理过,所以 fastQC 里基本没有问题,可以直接用来比对。不过这几天有一个数据比较异常,所以按照惯例地跑了一下 trim_galore,发现了一个意料之外的情况:
质控前,所有数据都有 Adapter Content 的警告,但是其他方面没有问题:
质控后,Adapter Content 的警告消失,但是 Per Base Sequence Content 全部爆红:
经过比较,原因都是 reads 末端的碱基组成异常导致:
经调查在 Trim Galore github 中已经有人提到过(issue #81),这里简单阐述下原因:
Trim Galore 在过滤时采用非常严格的策略,具体来说,它并不只在接头完全匹配时才进行移除,而是在末端开始进行逐个匹配。例如,假设识别到的接头序列为 AGATCGGAAGAGC,而某一个 reads 从末端开始为 ACCTCG,虽然这里从第二个碱基开始就不能和接头匹配上,但是由于第一个碱基 A 和接头相匹配,因此被 Trim Galore 移除,这也是为什么能在运行 trim_gal ...

基于 genmap 计算基因组 mappability
基因组的 mappability 是指测序产生的短读长能够被唯一且准确比对到参考基因组特定区域的能力。它反映了基因组不同区域因序列特征(如重复序列、低复杂度区域等)对比对结果可靠性的影响。例如,高度重复区域(如 Alu 元件)的 mappability 通常较低,因为 reads 可能匹配到多个位置。
在实际分析中,有时我们会过滤低 mappability 区域的突变,避免因比对错误导致的假阳性误判。本文将介绍如何使用生物信息学工具 GenMap 获得基因组的 mappability 信息。
GenMapGenMap github page:
https://github.com/cpockrandt/genmap
GenMap paper:
GenMap: ultra-fast computation of genome mappability
https://doi.org/10.1093/bioinformatics/btaa222
相较于先前的 mappability 计算工具(例如 GEM),GenMap 的搜索策略有所优化并且算法不依赖于启发式方法,因此具有更高的运行效率和 ...
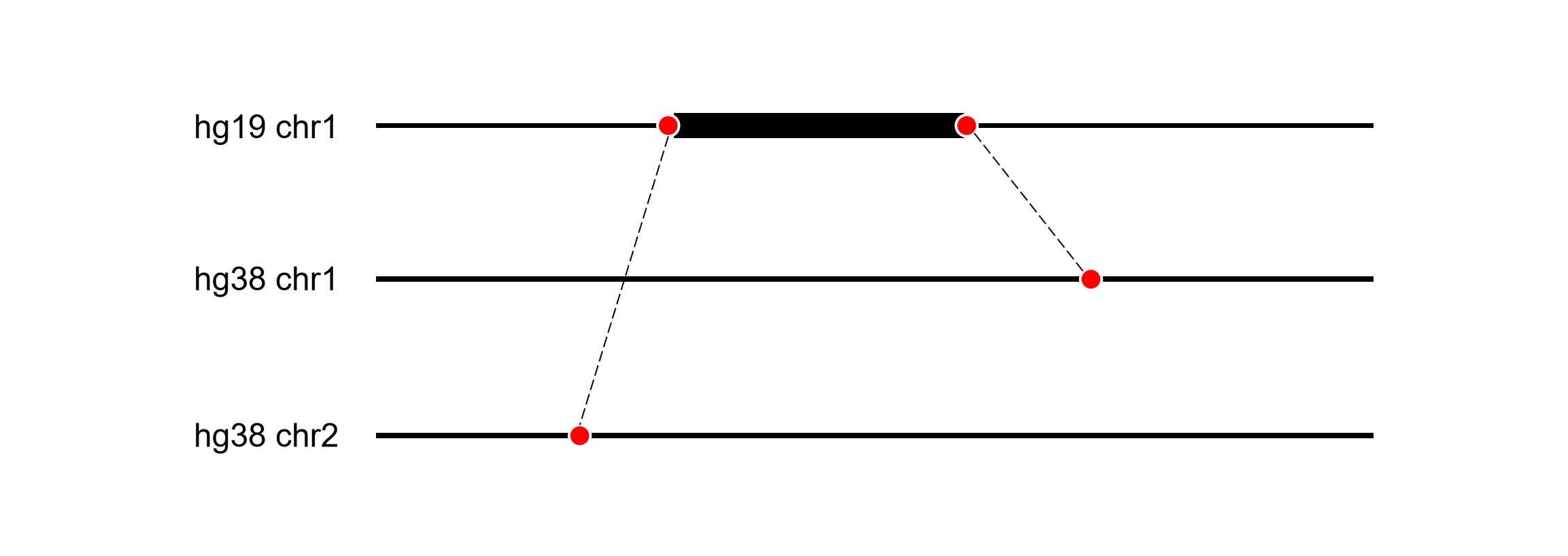
VCF 文件转坐标的方法汇总及注意事项
历史数据的整合分析往往涉及不同时期使用不同参考基因组版本产生的数据。例如,某些较早的研究可能基于 hg19/GRCh37 进行分析,而新的研究多采用 hg38/GRCh38。为了进行比较分析,需要将这些数据统一到同一个版本。
坐标转换是通过 chain file 来实现的,该文件记录了两个基因组版本之间的对应关系,包含了基因组重排、插入和删除等信息。转换工具会根据这些信息将原始坐标映射到目标版本的相应位置。
正文本文将介绍如何使用两款主流工具 CrossMap 和 GATK LiftoverVcf 以及最近的一个新工具 BCFtools liftover 进行 VCF 文件的坐标转换。在说明之前,首先简要介绍三个软件的差别:
CrossMap 除了 VCF 的坐标映射外,也支持很多其他文件格式的坐标转换。对于 VCF 文件而言,CrossMap 并不要求其具有完备的 Meta-information 和 Header line 部分,只需要突变数据行符合格式即可。
GATK LiftoverVcf 则主要支持对 VCF 文件的坐标进行转换。其要求 VCF 文件具有完整的 Meta-i ...

bedtools merge 不合并相邻区间(仅合并重叠区间)
关于 bedtools merge 的详细参数设定可见:
bedtools merge documentation:
https://bedtools.readthedocs.io/en/latest/content/tools/merge.html
默认情况下,bedtools merge 会对所有重叠区间及相邻区间进行合并,例如:
12345# 运行前chr1 10000 10001chr1 10001 10002# 运行后chr1 10000 10002
但在最近遇到的分析场景中,我仅想合并重叠的点并对它们的值求平均,并不想将它们与相邻的点合并起来,例如:
123chr1 10000 10001 0.1chr1 10000 10001 0.2chr1 10001 10002 0.3
这里我仅想在 chr1:10000-10001 上进行合并且求平均,但此时使用 bedtools merge -c 4 -o mean 会输出以下结果:
1chr1 10000 10002 0.2
这并不符合我的预期工作情况,在查阅参数说明后,发现了以下关键参数:
-d:Ma ...
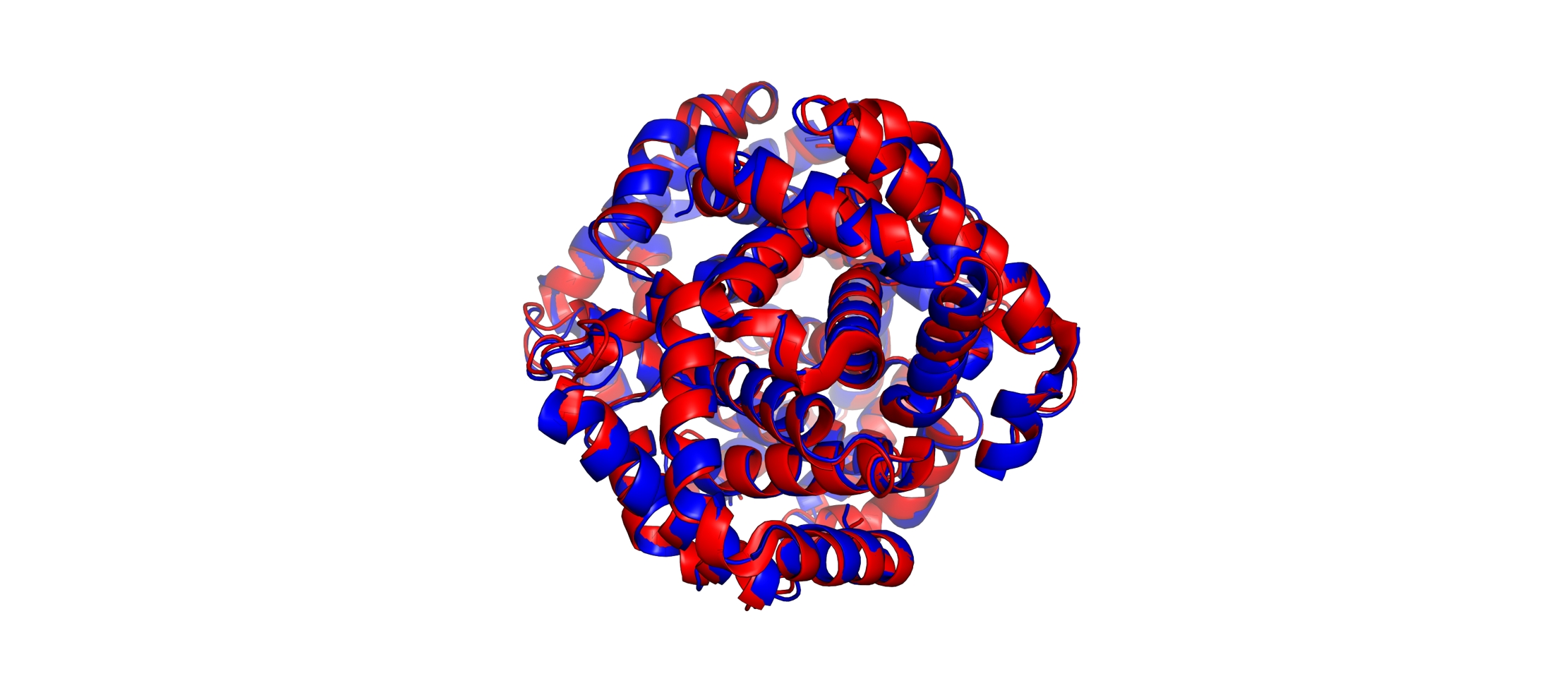
基于 US-align 计算两个蛋白结构之间的 RMSD 和 TM-score
在蛋白质结构研究里,比较不同蛋白质结构之间的相似性是一个核心任务,尤其是在结构预测、同源建模和结构功能关系研究中。以下是一些常用到的衡量蛋白结构间相似性的指标:
RMSD(均方根偏差):该值是衡量两种蛋白质三维结构之间原子位置差异的标准度量。较小的 RMSD 值意味着两个结构相似度较高,RMSD 值对大分子在全局对齐时的大小和形状变化非常敏感。
TM-score:相较于 RMSD,TM-score 更加关注结构的全局相似性,且对于蛋白质的尺寸和形状变化具有更高的鲁棒性。TM-score 的值介于 0 到 1 之间,值越接近 1,表示两个结构越相似。
本文将介绍如何使用 US-align 对两个蛋白结构进行 align(对齐)并计算其 RMSD 及 TM-score。
US-alignUS-align github page:
https://github.com/pylelab/USalign
该软件的研究团队也同样是 TM-align 的开发者。相比其他的计算工具,US-align 有以下优点:
支持比对除蛋白质以外的生物大分子及其复合物(例如 DNA / RNA 等)。
通过 ...
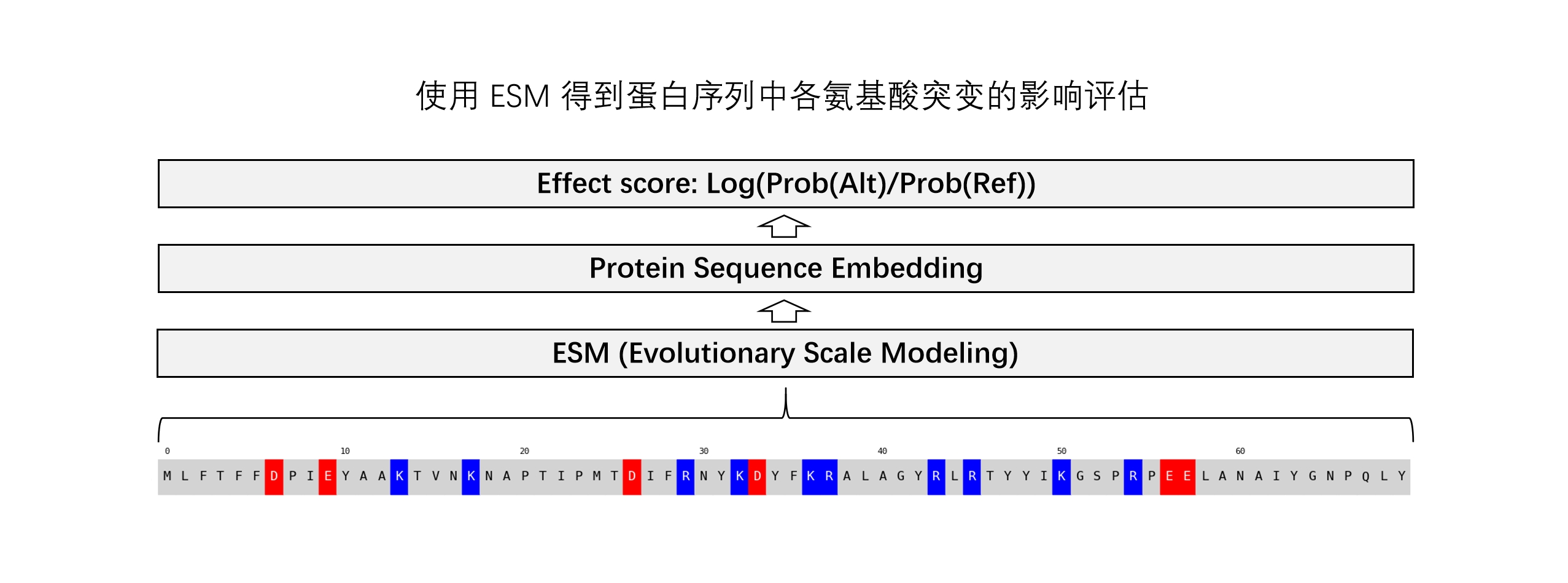
使用 ESM 计算蛋白序列的突变影响分数
ESM(Evolutionary Scale Modeling) 是由 Facebook AI Research 开发的蛋白质语言模型。其基于大规模的蛋白质序列数据进行训练,能够捕捉蛋白质序列中的复杂模式和进化信息,从而在多个生物学下游应用中表现出色。
预训练好的 ESM 可用于多个生物学下游应用,本文将就其突变影响分析方面的功能进行介绍。
关于 ESMESM 预测突变影响的方式是无监督的,也就是说 ESM 的训练过程中并未使用到与突变影响相关的标签进行有监督学习。其评估每个氨基酸错义突变影响程度的方式是掩蔽该位置上的氨基酸并计算两种氨基酸(参考氨基酸与突变氨基酸)在该位置出现的对数似然比:
ESM 的预训练阶段使用了类似于 BERT 的掩码任务,因此 ESM 可以给出每个位点里各类氨基酸出现的概率,而参考氨基酸(Ref,即未突变时的氨基酸)和突变氨基酸(Alt)的概率差异即可作为一种 effect score。可以简单理解为,突变氨基酸出现的概率比参考氨基酸低越多,说明这类突变越有害。这种无监督的评估方式也被用于许多其他的深度学习工具,例如 EVE 和 GPN 等。
Refe ...
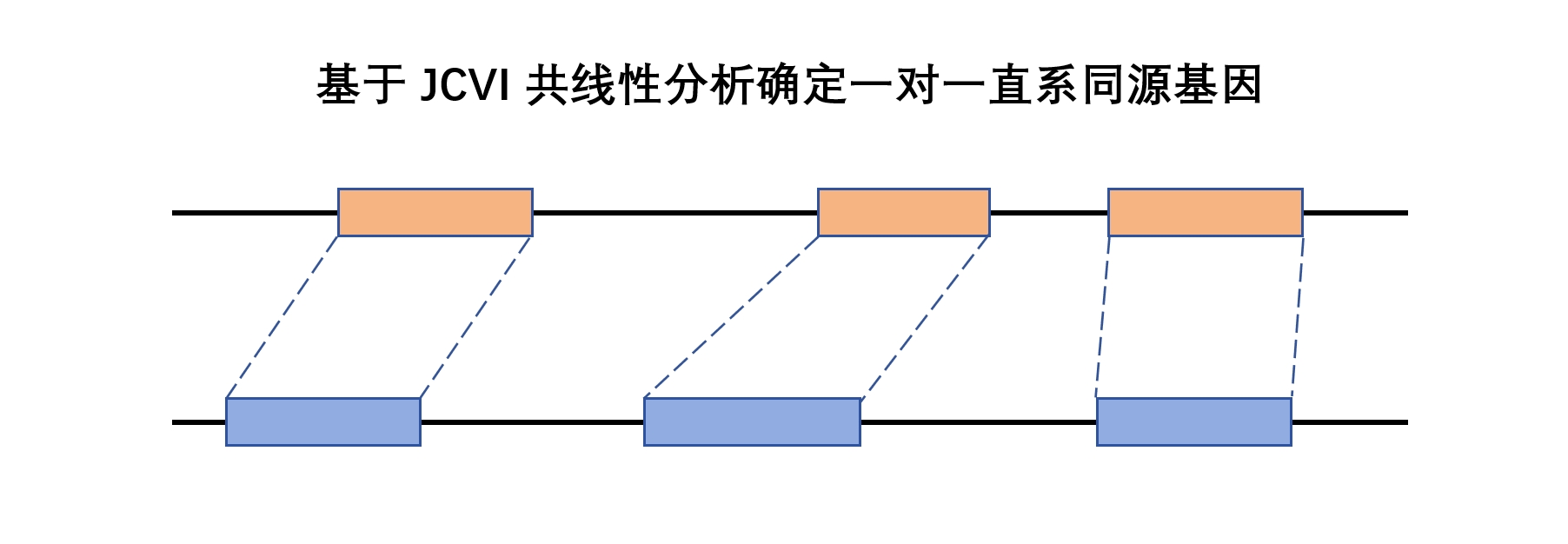
基于 JCVI 共线性分析确定一对一直系同源基因
前段时间因为课题需求,需要获取两个物种间的所有一对一直系同源基因对(One-to-One ortholog, 为避免文章内容冗余后面统称 OTO)。由于两个目标物种在 Ensembl 上都有,所以可以直接通过 BioMart 下载其基因的同源关系并提取出来。
但经过筛选和过滤后,发现 BioMart 中能确定的 OTO 数量极低,不太能满足分析需求。考虑到我所分析的目标基因类型在不同物种间具有高顺序保守性和序列保守性,因此想看看能不能通过 JCVI 的共线性结果来获取更多的 OTO 作为补充,发现效果还挺不错,固有此文。
JCVI 介绍JCVI 是一个用于比较基因组学分析的多功能工具包,共线性分析只是它其中的一个子功能。
JCVI 的安装,最简单的方式是通过 pip:
1pip install jcvi
也可通过 conda 或 mamba 安装:
1conda install bioconda::jcvi
此外也需要安装用于比对的 LAST:
1conda install bioconda::last
关于 JCVI 的运行,更多细节可见:JCVI tutorial
请注意, ...