
JuseKit(八) —— 计算转录组组装指标
更新变动及进度
JuseKit 故事传之开学第一天,我在课题组实验室搞新功能。
已有功能的相关教程请见:Juseの软件开发
本文所涉及功能借鉴了 Trinity 脚本的输出格式。
本次更新变动
- 完善了部分报错提示。
- 完善了部分文本显示。
- 新增了转录组组装指标计算功能。
目前的功能进度
- 提取最长转录本。
- 根据 id 提取序列。
- 对序列的 id 进行各种处理。
- 串联序列并得到分区信息。
- 批量进行序列格式转换。
- 批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。
- 批量进行序列的物种数和长度过滤。
- 火山图绘制。
- 气泡图绘制。
- 组装指标计算。
叠盾警告?:本软件解释权归属 Juse 所有,本软件能走多远具体得看 Juse 能坚持多久。
下载地址:https://github.com/JuseTiZ/JuseKit/releases
转录组组装指标计算
本文主要着重于这次更新新增的功能,其他模块请走这里。
要求的数据
需要输入的数据为 fasta 格式的序列文件。
可通过将文件拖拽至文本框或者点击右侧按钮读取文件路径。
操作示例:

结果文件会产生在序列文件的同一路径下,文件名为 assem_qc.txt。
目前返回的信息包括:
- 基因数量
- 最大长度、最小长度和平均长度
- Nxx 值
- GC 含量
以后视情况会继续完善这一功能,例如补充其他可以反应转录组组装质量的指标。
进阶应用
这一部分就相当于取最长转录本,关于该概念可见以往文章。
此处我将使用 _seq 作为基因标识进行(老版 Trinity 的序列格式):
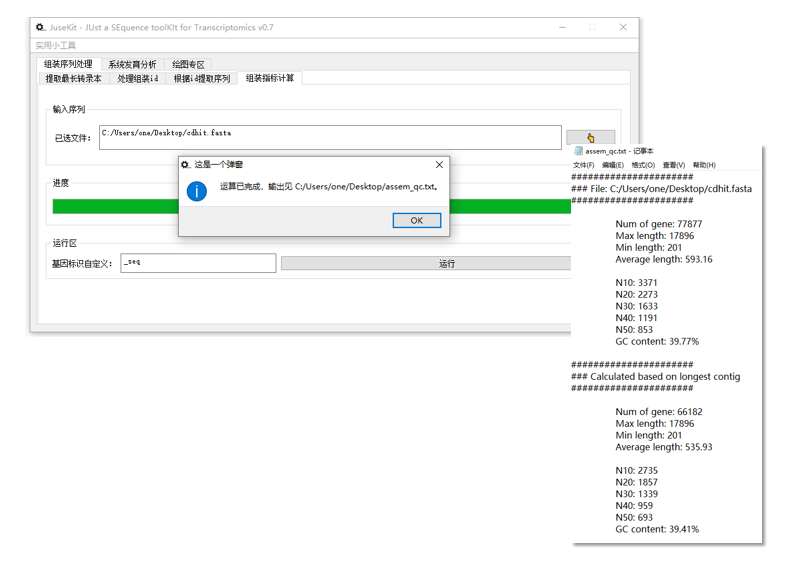
在指定基因标识后 JuseKit 会将每个基因的最长 Contig 作为其代表序列进行各指标计算。
如果在使用前后指标没有变化则说明该标识并不存在或者输入错误。
后记
后记?这次啊,没有后记!
最后是复制黏贴:
我会争取将这些功能慢慢完善,让它成为一个具有更广适用性的软件,希望能够帮助到某些盆友,当然我个人认为最大的可能是自娱自乐。
如果这个软件帮助到您了,您可以给它一个小小的 Star 聊表支持,或者在您汇报的时候引一下 https://github.com/JuseTiZ/JuseKit/ ,想必看着还是非常高端大气上档次的。
本博客所有文章除特别声明外,均采用 CC BY-NC-SA 4.0 许可协议。转载请注明来自 Juse's Blog!
相关推荐

评论