

JuseKit(五) —— 用于系统发育分析的序列过滤
generatePortalLinks(5);
更新变动及进度貌似距离上次更新已经有一段时日了,这一段时间忙完了各种事儿,也算迎来了本科生涯的最后一段悠闲时光。正好趁着这股兴致,把新的 Jusekit 文章给肝出来。
已有功能的相关教程请见:Juseの软件开发
Python 代码实现:比较转录组分析(七)—— 系统发育分析
本次更新变动
新增序列过滤功能。
序列串联输出的 log 文件新增 gap 信息。
新增了部分按钮选择文件时的后缀选项。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(已实现)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(已实现)
批量改后缀。(已实现)
批量进行序列格式转换。(已实现)
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。(已实现)
批量进行序列的物种数和长度过滤。(已实现)
叠盾警告?:本软件解释权归属 Juse 所有,本软件能走多远具体得看 Juse 能坚持多久。
下载地址:https://github.com/JuseTiZ/JuseKit/releas ...
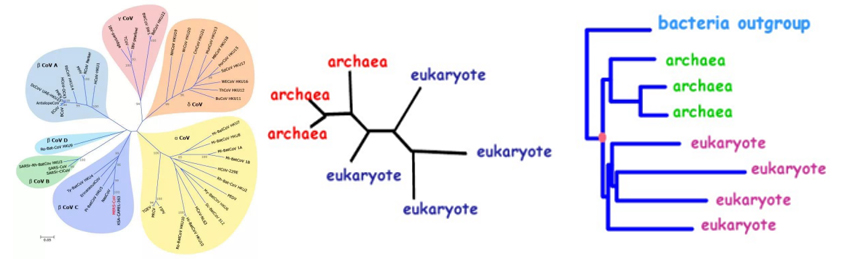
关于 PAML 的一二三事
PAML 简介PAML,全称 Phylogenetic Analysis by Maximum Likelihood,由国人大佬杨子恒教授开发,是一款用于进行各种系统发育分析的软件包。里面的诸多模型可以说是系统发育学文章里的常客,其所涵盖的功能包括但不限于:
重建系统发育树。
检测选择压力。
估计进化速率。
估计物种或基因的分歧时间。
想要最详细地了解这个软件,可直接下载官方 tutorial 进行啃读:
PAML Manual:http://abacus.gene.ucl.ac.uk/software/pamlDOC.pdf
PAML FAQs:http://abacus.gene.ucl.ac.uk/software/pamlFAQs.pdf
也有其他博主写的精细介绍,详见:PAML-discussion-group from 简书
如果有些报错问题或者运行问题在网上找不到解释,也许可以看看 PAML 的谷歌论坛。
提示,本文将基于 linux 系统的操作进行讲解。
更新日志2023.05.20 补充了支位点模型的局限性。
2023.06.21 补充了自由比模 ...
JuseKit(四) —— 序列格式转换以及 Orthogroup 的 cds 提取
generatePortalLinks(4);
更新变动及进度已有功能的相关教程请见:Juseの软件开发
本次更新变动
多种序列格式的转换(包括一些常见的 nex phylip 等)。
将 PEP 序列转变成 CDS 序列。
新添了一个学习计时器。
修复了部分 Bug。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(已实现)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(已实现)
批量改后缀。(已实现)
批量进行序列格式转换。(已实现)
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。(已实现)
批量进行序列的物种数和长度过滤。(拟下一次更新)
增加自动绘图模块(例如火山图、富集图等)。(考虑中,新的模块将会大幅增加软件大小,Juse 正在权衡利弊)
增加各种系统发育分析辅助工具(例如批量计算 RF 值或进行 ILS test Introgression detection 等)。(考虑中,新的模块将会大幅增加软件大小,Juse 正在权衡利弊)
叠盾警告?:不保证这些功能和想法一定会实现,本软件 ...
JuseKit(三) —— 串联序列、根据id提取序列、批量修改文件尾缀
generatePortalLinks(3);
更新变动及进度已有功能的相关教程请见:Juseの软件开发
更新日志2023.06.21 完善了输出的 log 信息。
本次更新变动
对序列进行串联(支持 IQ-TREE 分区文件的生成)。
根据 id 提取序列。
批量修改文件的后缀(已经补充在第二篇的进阶应用中了)。
修复了部分 Bug。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(已实现)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(已实现)
批量改后缀。(已实现)
批量进行序列格式转换。(拟下一次更新)
批量 RBH 得到一对一直系同源基因。(拟废弃,因为 diamond 的 windows 部署有些麻烦)
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。(拟下下次更新)
增加自动绘图模块(例如火山图、富集图等)。(考虑中,新的模块将会大幅增加软件大小,Juse 正在权衡利弊)
叠盾警告?:不保证这些功能和想法一定会实现,本软件解释权归属 Juse 所有,本软件能走多远具体得看 Ju ...
JuseKit(二) —— 序列id简化、加前缀尾缀或转变为物种名
generatePortalLinks(2);
更新变动及进度本次更新变动有:
对序列的 id 进行前缀或后缀(尾缀)添加。
对序列的 id 进行简化。
对序列的 id 进行转换(变为物种名)。
部分功能的 Python 代码实现:比较转录组分析(三)—— 组装的质量检测与去冗余
更新变动2023.04.13 新增了批量替换尾缀的功能,在进阶应用里进行了补充。
2023.04.24 处理组装 id 中的 删除原有文件 功能在什么时候都能使用了。
目前的功能进度
提取最长转录本。(已实现)
根据 id 提取序列。(拟下一次更新)
对序列的 id 进行各种处理。(已实现)
串联序列并得到分区信息。(拟下一次更新)
批量 RBH 得到一对一直系同源基因。
新的想法
批量提取 Orthofinder 的 orthogroup 对应的 CDS 序列。
批量进行序列格式转换。
批量改后缀。(拟下一次更新)
叠盾警告?:不保证这些功能和想法一定会实现,本软件解释权归属 Juse 所有,本软件能走多远具体得看 Juse 能坚持多久。
下载地址:https://github.com/J ...
JuseKit(一) —— 提取最长转录本
generatePortalLinks(1);
前言JUst a SEquence toolKIt for Transcriptomics
不同于其他的生信软件,该软件将着重于一些非常基本的功能,并且以全中文进行编写。
已有的生信软件一直以来只能用一个简单且粗暴的词来形容:高端大气上档次。如今,我将亲自定义简陋朴素接地气。
—— Juse
这篇文分享第一个基本功能:提取最长转录本。
下载地址:https://github.com/JuseTiZ/JuseKit/releases
Python 代码实现:比较转录组分析(三)—— 组装的质量检测与去冗余
提取最长转录本它目前只有那么一个简陋的页面:
首先,默认情况下它提取 Trinity 组装中的最长转录本,因此基因标识锁定为 _i,后文将提到怎么活用基因标识。
输入 fasta 的方法有两个,一个是通过文本框直接输入,一个是读取文件,后者优先级更高。
输出文件的名称默认是 longest_contig.fasta ,可以自定义修改。注意:
文件的路径和输入文件一致。当通过文本框直接输入时,它将会在 .exe 所在文件 ...

系统发育转录组学是否靠谱?
前言我一直认为,阅读文献应该是带着明确的目的去读,要么是想要从中学习到什么东西,要么是想通过它解答一些问题。
作为开篇,我先来分享一下对我正在研究的东西有支持性的文章:
Is Phylotranscriptomics as Reliable as Phylogenomics?
from Mol Biol Evol
事实上,很久之前我认为用转录组找出来的 ortholog 并不可靠,里面有太多的不确定因素会干扰结果的准确性,这也是它和基因组最大的区别所在。所以我有一段时间一度在怀疑自己这么做到底是不是对的(菜鸟就是年轻气盛啊!)。
引言首先,研究者肯定了基因组在系统发育分析中的优越性,但也指出其成本依然昂贵,相比之下相当便宜的转录组已经成为更多研究者的选择,不过转录组依然具有以下缺点:
基因表达在组织间具有差异性(例如脑组织和肌肉组织所表达的基因可能有所不同)。
基因表达在物种间具有差异性(例如不同物种的同一基因可能具有不同的表达)。
高表达基因往往进化较为缓慢,而这一类基因在转录组中是大量出现的(也有其他观点认为相较于整个基因组背景而言这些编码序列进化速度更快,因为可能包括正在快 ...

关于 mamba install 时会出现的两个错误解决方案
工欲善其事必先利其器篇堂堂连载!这篇文分享一下使用 mamba 进行安装时遇到报错该怎么解决。
一般来说错误有这么两个:
File not valid: file size doesn't match expectation
RuntimeError: Multi-download failed.
这两错误在 mamba github 上已经有人提出来了,并且指出了明确的报错原因:源有问题。
File not valid: file size doesn't match expectation 时说明源网站没有正确识别 mamba 并返回了错误的信息,例如清华的镜像源会把 mamba 当成手机设备因此会先返回一个 "用移动设备下载会消耗过多流量建议改用计算机" 的劝告信息。
RuntimeError: Multi-download failed. 时说明源可能已经不可用,例如中科大的 conda 镜像源已经废弃。此外也有可能是因为配置的源拼写有问题,例如将 conda 误拼成 condo。
因此解决办法也是很简单的,那就是把出现问题的镜像源删了,或者是修改成可以使用的不会报错的镜像 ...
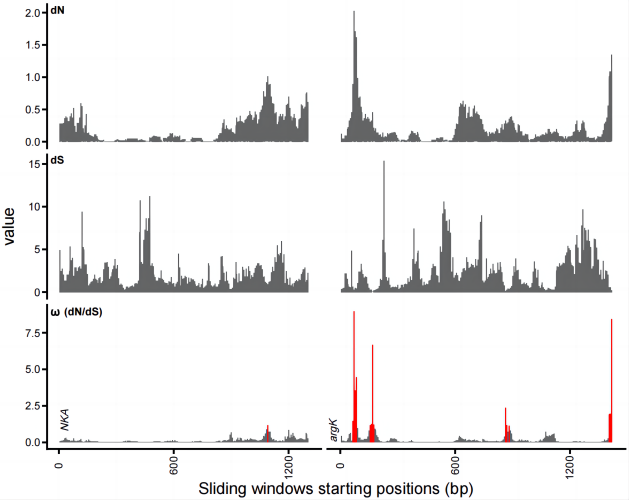
单基因 dN、dS 和 ω 的滑动窗口绘制方法
需要准备的文件和软件需要提前准备好的:
用于进行滑动窗口绘制的基因比对文件(核苷酸、密码子比对格式)
KaKs_Calculator 3.0
Python
R
更新日志2023.07.03 解封文章,简化了部分内容。
滑动窗口绘制过程计算滑动窗口将需要进行滑动窗口计算的 fasta 文件放置于某个特定文件夹中。
然后使用下述 Python 脚本:
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130131132133134135136137138import sysimpo ...
当 cmake 遇上 permission denied
工欲善其事必先利其器,软件就是生信人的武器,这篇文就是分享使用 cmake 配置遇到 permission denied 时应该怎么解决。
一般错误出现的情况一般来说,cmake 安装软件的流程都是大致如下:
1234$ git clone xxx # 下载某个软件$ cd xxx # 进入下载的文件夹中$ cmake . # cmake 进行编译配置$ make install # make 进行安装
在最后一步 make install 的时候,有时会出现 permission denied。
如果具有 sudo 权限这种情况一般发生在自己的电脑上(root 用户),又或者拥有 sudo 权限,加一个 sudo 即可。
1$ sudo make install
如果不具有 sudo 权限本篇文章的重心,permission denied 的原因是自己没有在 /usr/local 配置文件的权限而又无法通过 sudo 解决,这个路径是很多软件默认的安装路径,但同样也是可选的,所以解决方法就是把安装路径改到自己有权限的路径即可。
123# cmake 时指定其他安装路径$ cmak ...
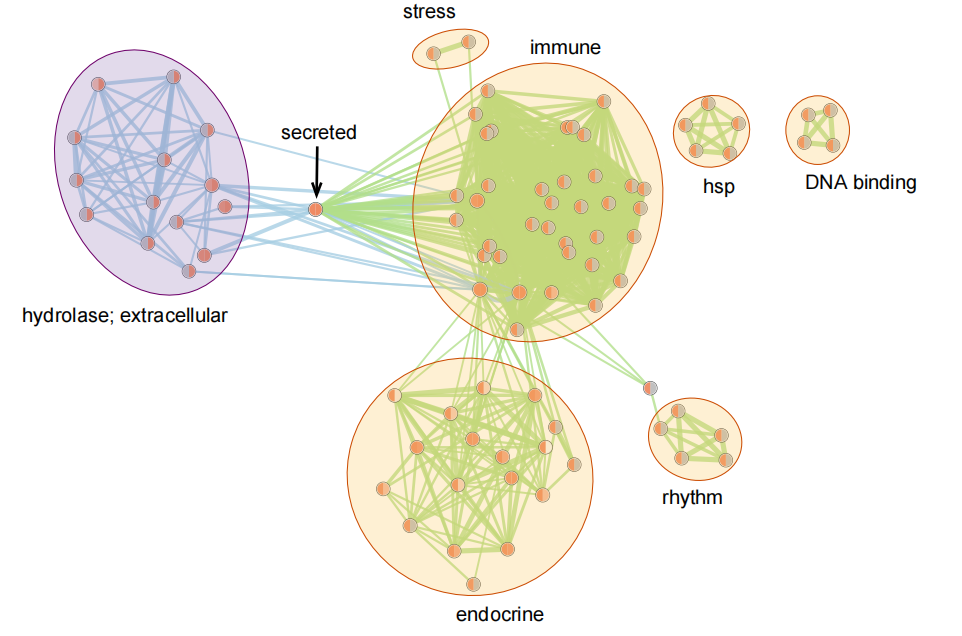
基于 Cytoscape 的富集网络图绘制
所需文件需要准备的文件:通过 David 得到的富集分析结果表格。
软件的下载方式请自寻,请支持正版。
注意,本文着重的是无参转录组富集分析结果的网络图绘制,如果是有参基因组请移步它处。
绘图过程首先我们需要通过 David 分析得到富集分析结果文件。
DAVID 富集分析教程可见:比较转录组分析(六)。
注意,这里对我来说仅选择 GO 的结果是不够的,因为这样会导致富集网络图中的节点数量过少(因为富集的 GO term 过少),因此我选择了更多的项(DAVID 的所有默认项)来进行富集结果的输出,可以根据个人的情况进行相应的调整。
得到表格以后可以直接输入 Cytoscape 进行绘制。
首先在 Apps 一栏中选择 App Manager 并下载以下两个插件:
Enrichment Map
WordCloud
此后打开 Apps 中的 Enrichment Map ,会跳出一个窗口,操作步骤如下图所示:
在这里可以输入多个表格(例如分别输入上调基因和下调基因的富集分析表格)。此外,在输入了表达量文件后左下角部分还可对基因进行表达量过滤,右下角可以通过调整参数来调节节点的聚集 ...

比较转录组分析(七)—— 系统发育分析
generatePortalLinks(7);
关于分类学和系统发育分析分类学,这个学科虽然听起来很简单,但从整个生物史的尺度出发都能算的上是一个非常具有深度且一直以来都争议颇多的学问。从几个世纪前,分类学家就已经致力于通过各种形态学差异或者生活史差异来描述不同物种之间的亲缘关系并且把它们分门别类成不同的类群。在那个时候,不同的学者也会根据 “个人之见” 进行颇具主观性的划分(而且某些近缘物种之间的形态学差异确实可以迥然不同),因此许多分类单元并不统一且一直以来都在不停变动,这一些现象在达尔文先生的《物种起源》中表现得淋漓尽致。
即使古早的分类方法并不完全准确,分类学家依然完成了非常瞩目的成就,就拿现在的物种分类情况来说,很多框架依然与以前一致,并且新的分类单元也是基于已有的框架延申出来的。
测序手段的发展算得上是分类学的一个里程碑,从分子层面的观察让我们对于物种的分类关系有了更为准确的把握,这催生了大量分类错误的纠正以及新物种的发现,并且由此诞生出来了分子系统发育学这一学科。目前很多生信软件都能帮助我们探讨不同物种间的系统发育关系。
这篇文将分享如何通过一些软件来进行系统 ...
用于 IQtree 的序列串联方法
前言因为这一段时间很懒而且现实生活的事情又多,所以系统发育分析的正式文章教程可能要搁置一会,但是想着总得更新些东西出来,所以就以这篇文章作为正餐开始前的一叠小菜分享分享。
事实上序列串联对于刚涉及到这一块的新手来说确实也会有些迷糊,比如说串联是怎么个串联法,需要准备什么文件,文件的内容格式应该是怎样等等问题都会存在,这篇文里就针对这一点讲一讲。
更新日志2023-04-07 简化了脚本,并完善了脚本的注释。
2023-07-04 完善了脚本,并添加了部分内容。
需要知道的知识一些系统发育分析中的概念是最基础的,如果未作了解或了解不深可以看一下这篇文章:生命之树及其应用 。写的非常全面和具体,里面涉及到的各种术语也很专业,同时也介绍了很多常用的软件等。本文涉及到的地方就是这篇文章展望部分中第一点讲的 “超大树构建方法的革新” 中的超矩阵方法。
序列串联方法需要准备的文件首先按照惯例推出几个能够实现的软件:Phylosuite、FASconCAT-G。前者用起来非常方便但是我只下了 windows 的版本,虽然它有 linux 的版本但我也没去调查怎么用,索性就自己写了个脚本 ...
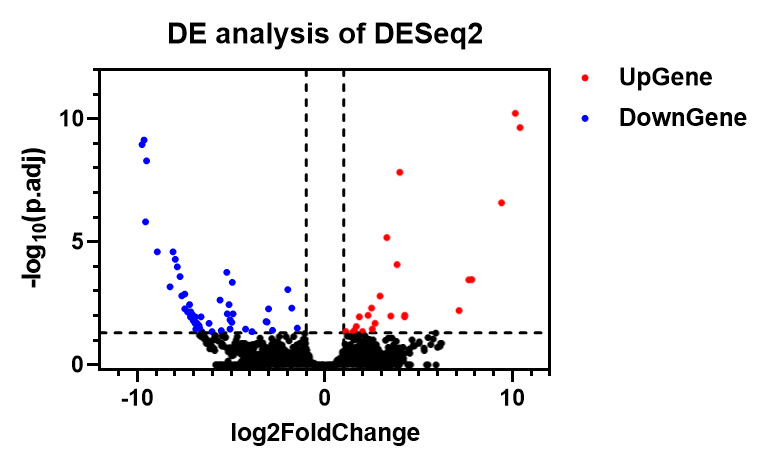
基于 GraphPad Prism 的火山图绘制
软件信息此文中使用的 GraphPad Prism 版本为 9.4.1,获取方式请自寻,请支持正版。
需要准备的文件:差异表达分析结果表格。
9.0 以上版本的 GraphPad Prism 已经支持火山图绘制,但是绘图效果并没有利用散点图绘制来的好,因此这篇文还是通过散点图的方式来实现火山图绘制,如果想要试试 Prism 自带的火山图可以在点开 Prism 时选择 Graph Portfolio 里的 Volcano Plot 进行:
更新日志2023.07.07 精简了内容,增加了题外话。
绘图过程在进行绘图之前,需要先筛选出 |log2FC| > 1 以及 adjustp < 0.05 的点(可以选用不同的阈值),然后将它们的 log2FC 放在新的一列,最后效果应该如下所示:
具体的实现过程可以通过 Python 脚本或者 R 进行,处理以后的表格用来绘制火山图。
点进 GraphPad Prism 后,选择最上方的 XY,各项选择如图:
生成表格后,导入差异表达分析结果(复制黏贴整个表格即可),此后删掉除了所需要的数据以外的列,最后如下图:
其中第一 ...