bismark 分析 Bisulfite-Seq 数据的流程示例
前言
以下是相关的官网链接和参考资料,如果想要得到除本文内容以外更全面深入的了解,建议直接跳转相关链接进行查阅:
Bismark github:
https://github.com/FelixKrueger/Bismark
Bismark documentation:
https://felixkrueger.github.io/Bismark/
本文比对及去重等流程均参考自 Bismark documentation
此外,以下内容在本文中不会涉及到,有需要请自行探索:
- 数据的质控
- 除 Bismark 以外其他软件的安装
通过以下流程可以得到:
- 基因组上 CpG 位点的甲基化信息
请阅读完全文后再进行相关分析,建议先使用 bismark 的示例数据观察相关命令是否存在问题。
正文
下载最新版本的 bismark(至 2024/04/19):
1 | wget https://github.com/FelixKrueger/Bismark/archive/refs/tags/v0.24.2.tar.gz |
也可前往 github release 下载指定版本或可能已存在的更新版本:
https://github.com/FelixKrueger/Bismark/releases
下载后解压并将对应文件夹置入环境变量:
1 | tar xzf Bismark-0.24.2.tar.gz |
由于 bismark 中比对等操作均由 bismark 本身调用比对软件进行,因此在运行后续代码前,请确保正确版本的 Bowtie2 或 HISAT2 已经被置入环境变量中,如果想自行指定,可在后续执行时添加以下参数:
1 | --path_to_bowtie2 </../../bowtie2> or |
bismark 默认使用 bowtie2,除非特别指定 --hisat2。
比对
比对前,使用 bismark 构建基因组 C>T G>A 的索引:
1 | bismark_genome_preparation --verbose --parallel [thread] [path] |
path换为基因组序列文件储存的路径,bismark 将自动识别.fa及.fasta文件并建立索引(压缩格式.gz也能识别)。thread处填入使用的线程数量,注意最终将会使用2 * thread个线程。
在构建完索引后,就可以通过 bismark 对数据进行比对,在比对前,建议先记录版本信息:
1 | bismark --version > bismark.version 2>&1 |
比对命令(单端):
1 | bismark -L [seed length] -N [mismatch number] --genome [path] --parallel [thread] -o [output_dir] [fastq] > [logfile] 2>&1 |
比对命令(双端):
1 | bismark -L [seed length] -N [mismatch number] --genome [path] --parallel [thread] -o [output_dir] -1 [fastq_R1] -2 [fastq_R2] > [logfile] 2>&1 |
相关参数解释:
seed length指定比对过程中使用的基础片段的长度(默认为 20),最大可设置为 32,该值越小时比对越慢,但同时也增加了召回率。该选项只在 Bowtie2 可用。mismatch number指定比对过程中基础片段允许的错配数量(默认为 0)。可设置为 1,设置为 1 时比对会减慢很多,但同样增加了召回率。该选项只在 Bowtie2 可用。thread指定并行的实例,注意其不是单纯的线程数,由于一个bismark进程就已经使用了多个核,以小鼠基因组为例,该值设置为 4 时将会使用大约 20 个核以及 40GB 左右的 RAM。因此请根据基因组大小、空闲的内存及核数量对该参数进行调整。path即构建索引时使用的path。output_dir为输出的目录,fastq指定测序数据文件,logfile记录比对过程中的屏幕输出。
注意!如果测序数据由 PBAT 建库得到,需要添加 --pbat 参数。此外,bismark 也提供了 --local 参数,指定此参数可以提高该文库类型的 map 比率,但是在 documentation 中 bismark 团队并不推荐这么做。
比对完成后,请先检查比对情况,相关的比对报告将储存在 output_dir 中以 _report.txt 结尾的文件里,如果是单端数据且使用 Bowtie2 比对,则相关报告文件将以 _bismark_bt2_SE_report.txt 结尾。打开文件,检查其中 Mapping efficiency 一项:
1 | Final Alignment report |
如果 Mapping efficiency 在可接受范围内,则可进行后续分析。若该值过低(例如不超过 30%),则需考虑更改过滤参数以提升该值。一个折中的选择是适当提升 seed length 并设置 mismatch number 为 1。
考虑到比对的用时都较长,因此在第一次比对时就应尽量根据实际情况决定一个好的参数起点,例如:
- 测序数据的 reads 长度?是单端还是双端?
- 测序数据的质量如何?
去重
基因组上相同位置的比对可能是由 PCR 扩增导致的,删除重复的 reads 有助于避免 PCR 引入的序列 bias 并提高后续相关分析的准确性。但请注意,不建议对目标富集型文库(target enrichment-type library)例如 RRBS 及 amplicon 等进行该操作,因为在这些类型的文库中,重复序列可能是有意义的。
1 | deduplicate_bismark --version > deduplicate_bismark.version 2>&1 |
1 | deduplicate_bismark --output_dir [output_dir] --outfile [prefix] [files] > [logfile] 2>&1 |
output_dir填入去重后的文件输出目录。prefix填入去重后的文件前缀名称。files填入需要去重的文件。logfile填入记录屏幕输出的日志文件。
如果此处想要将多个比对结果放在一起进行去重,则需指定 --multiple 参数,此时 bismark 将会把所有的输入文件视为单个样本连接在一起并进行去重。
最后的去重结果将在以 .deduplication_report.txt 结尾的文件中,该文件里将展示共有多少比对被移除。
统计甲基化信息
该步使用的 bismark_methylation_extractor 命令具有非常多的可选参数,建议通过 bismark_methylation_extractor --help 查看详细信息,此处给出个人使用的命令:
1 | bismark_methylation_extractor --version > bismark_methylation_extractor.version 2>&1 |
1 | bismark_methylation_extractor --gzip --bedGraph [bamfile] --output_dir [output_dir] --parallel [thread] > [logfile] 2>&1 |
bamfile 处填入经过上述处理后得到的最终 bam 文件。此处 --gzip 表示输出文件以 gzip 进行压缩,--bedgraph 使 bismark 输出 bedgraph 文件。也可不使用 --bedgraph 参数并通过结果文件自行处理生成。
通过上述命令将得到以下尾缀结尾的文件:
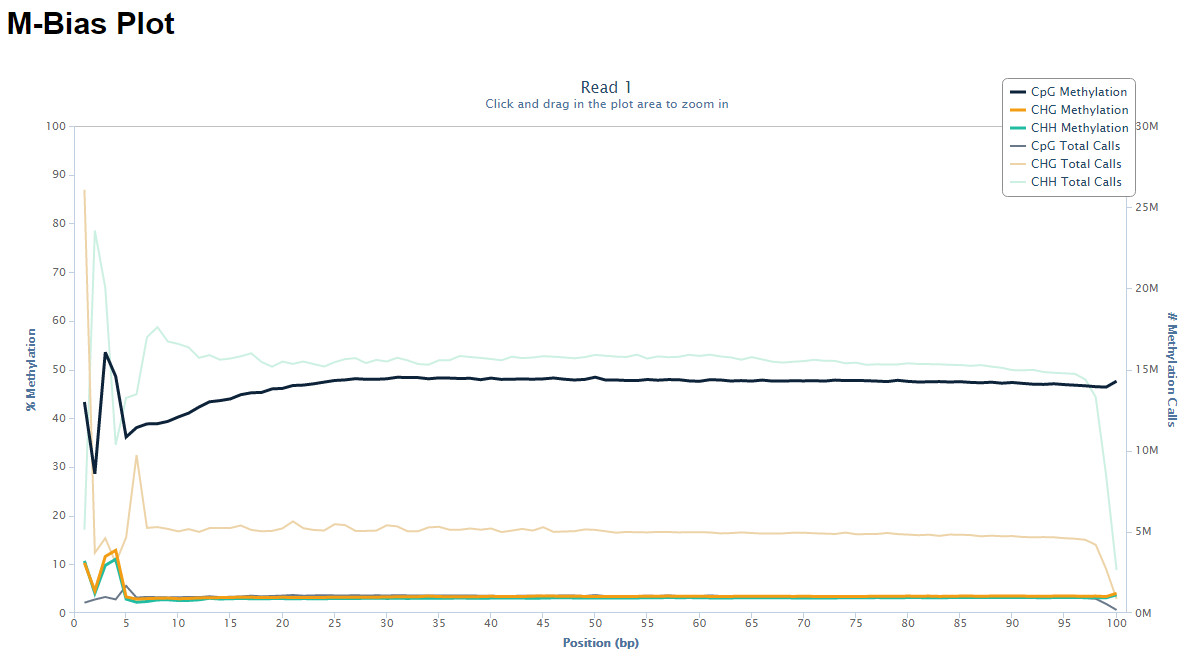
.bismark.cov.gz该文件共六列,分别代表:染色体、起始位点(1 坐标)、终止位点、甲基化比例、甲基化的 read count、非甲基化的 read count。.bedGraph.gz该文件共四列,分别代表:染色体、起始位点(0 坐标)、终止位点、甲基化比例。.M-bias.txt该文件展示了 reads 中每个 position 的平均甲基化水平,可通过画图可视化,偏离水平线可能表示存在 bias [Bock 2012, Nat Rev Genet]。_splitting_report.txt相关信息的统计汇总。
通过 .bismark.cov.gz 生成 .bedGraph.gz 的方式:
1 | zcat xxx.bismark.cov.gz | awk -F'\t' 'OFS="\t" {print $1, $2-1, $3, $4}' | gzip > xxx.bedGraph.gz |
此后可以通过 bedgraph 文件生成 bigWig 文件,请注意,由于 bedGraphToBigWig 并没法输入压缩文件,因此你可能需要对上述文件进行解压,或者不使用 gzip 进行压缩:
1 | bedGraphToBigWig xxx.bedGraph genome.chrom.sizes output.bw |
关于 chrom.sizes 文件的获取方式可参考之前的 ChIP-seq 文章。
以下是 bismark documentation 中给出的示例 M-bias plot(仅为示例,并不代表这种属于正常情况):
后记
bisulfite-seq 数据的分析也可以通过 gemBS Pipeline 直接进行,请注意该 Pipeline 目前仅支持双端数据。
也不必过多担心参数的选择,大多数情况下可能仅有 bismark 的 -L 和 -N 需要进行调整,如果不知道应该设置多少为宜建议统一使用默认参数,再根据结果进行修改。