
JuseKit(一) —— 提取最长转录本
前言
JUst a SEquence toolKIt for Transcriptomics
不同于其他的生信软件,该软件将着重于一些非常基本的功能,并且以全中文进行编写。
已有的生信软件一直以来只能用一个简单且粗暴的词来形容:高端大气上档次。如今,我将亲自定义简陋朴素接地气。
—— Juse
这篇文分享第一个基本功能:提取最长转录本。
下载地址:https://github.com/JuseTiZ/JuseKit/releases
Python 代码实现:比较转录组分析(三)—— 组装的质量检测与去冗余
提取最长转录本
它目前只有那么一个简陋的页面:
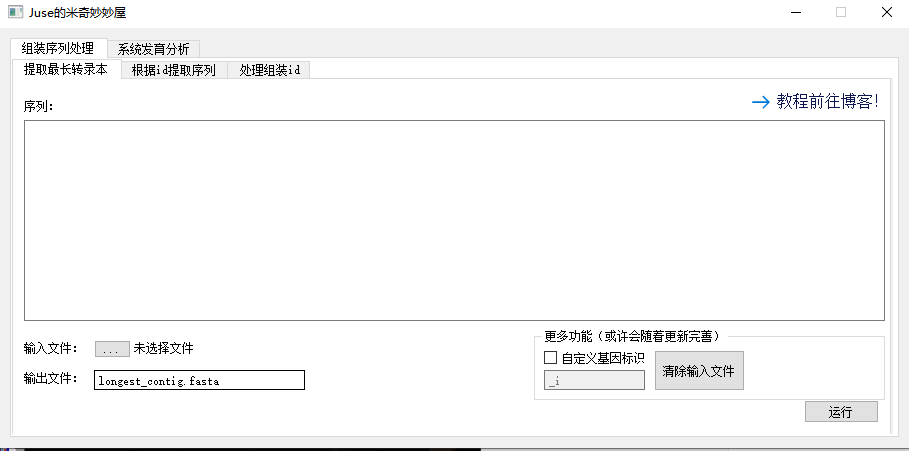
首先,默认情况下它提取 Trinity 组装中的最长转录本,因此基因标识锁定为 _i,后文将提到怎么活用基因标识。
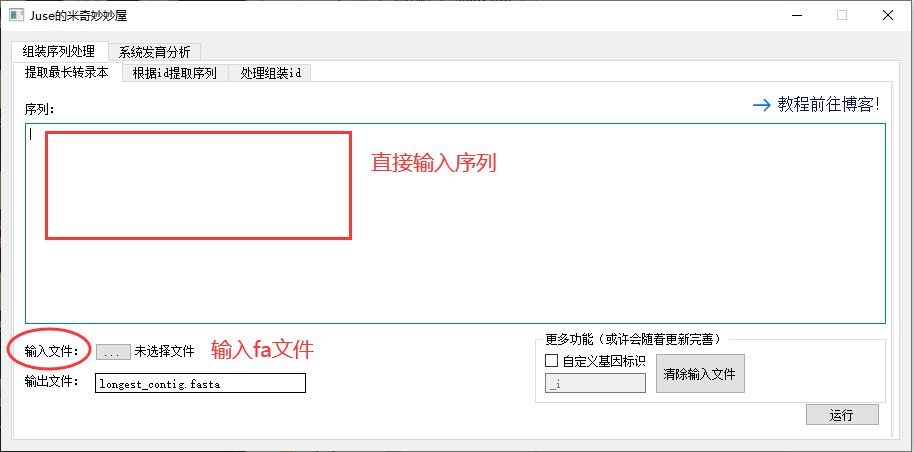
输入 fasta 的方法有两个,一个是通过文本框直接输入,一个是读取文件,后者优先级更高。
输出文件的名称默认是 longest_contig.fasta ,可以自定义修改。注意:
- 文件的路径和输入文件一致。当通过文本框直接输入时,它将会在
.exe所在文件夹下输出。 - 当通过文本框直接输入时,若存在报错则说明序列出现问题或者输出文件没填,此时输入的序列会存在于
.exe所在文件夹的tmp.fa中。 - 如果是通过文本框直接输入,那么一定要留意最后一个序列结尾是否有换行符,如果没有要加上。
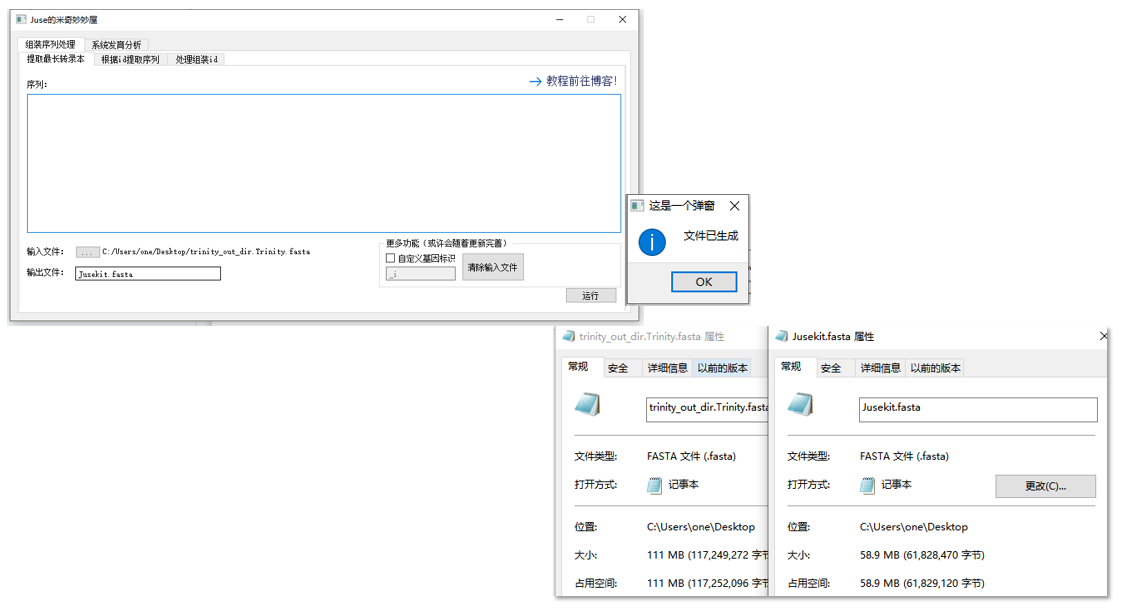
准备好后点击运行,将会产生对应的输出文件,实际操作:
进阶应用
自定义基因标识是什么意思?它有什么用?
默认提取的文件中,不同的 isoform 是长这样的:
1 | >TRINITY_DN16889_c0_g2_i1 |
这里,它们的基因部分是 TRINITY_DN16889_c0_g2 ,不同的 isoform 可以由 _i 标识。
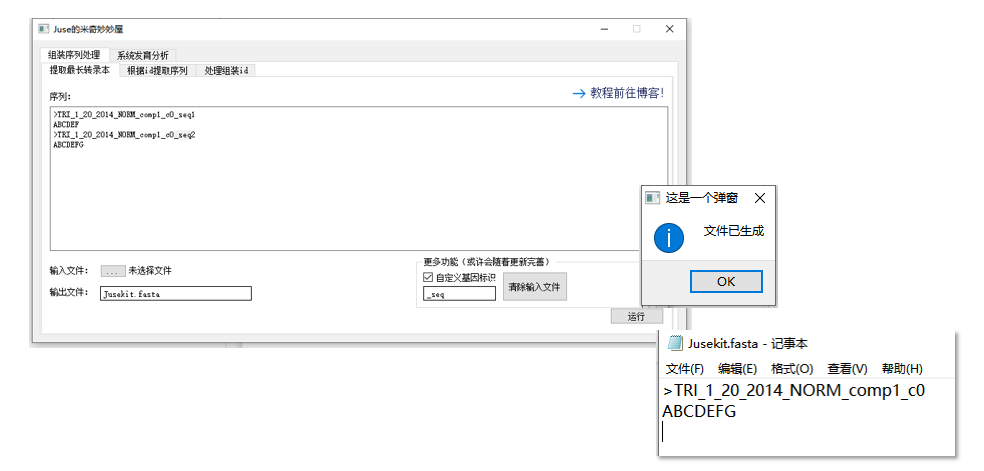
如果要提取其他的 “最长”,例如古早的 Trinity 组装版本:
1 | >TRI_1_20_2014_NORM_comp1_c0_seq1 |
可以看到,基因部分由 TRI_1_20_2014_NORM_comp1_c0 标识,不同 isoform 由 _seq 标识。
这时可以通过自定义基因标识,提取这种形式的最长转录本。
同理也适用于 Transdecoder 预测出来的最长蛋白(或 CDS)提取(.p)和 MitoFinder 跑出来的多条线粒体 contig(_mtDNA_contig)。
1 | # Contig 示例 |
后记
很显然,这个软件目前的大小根本没法匹配上它的功能,原因在于现在的大小主要来源于 PyQt5 的封装,也就是说现在框架已经搭好,但也仅有框架而没有内容。
我会争取将这些功能慢慢完善,让它成为一个具有更广适用性的软件,希望能够帮助到某些盆友,当然我个人认为最大的可能是自娱自乐。
如果这个软件帮助到您了,您可以给它一个小小的 Star 聊表支持,或者在您汇报的时候引一下 https://github.com/JuseTiZ/JuseKit/ ,想必看着还是非常高端大气上档次的。
