需要准备的文件和软件 需要提前准备好的:
用于进行滑动窗口绘制的基因比对文件(核苷酸、密码子比对格式 )
KaKs_Calculator 3.0
Python
R
更新日志 2023.07.03 解封文章,简化了部分内容。
滑动窗口绘制过程 计算滑动窗口 将需要进行滑动窗口计算的 fasta 文件放置于某个特定文件夹中。
然后使用下述 Python 脚本:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 import sysimport reimport osimport timeog_file = open (sys.argv[1 ],"r" ) fa_dir = os.path.abspath(sys.argv[2 ]) sli_len = int (sys.argv[3 ]) gap_len = int (sys.argv[4 ]) cpu_num = sys.argv[5 ] code_model = sys.argv[6 ] time_start = time.strftime('%Y%m%d-%H%M%S' ) os.mkdir(f'SWworkdir_{time_start} ' ) ogall = [] for line in og_file: oglist = [line.split('\t' )[0 ], line.split('\t' )[1 ].strip()] ogall.append(oglist) def readfa (fasta ): id_seq = {} with open (fasta, 'r' ) as fa: for line in fa: if line.startswith(">" ): idofseq = line.split()[0 ][1 :] id_seq[idofseq] = '' else : id_seq[idofseq] += line.strip() return id_seq def fa2axt (oglist, fasta, name ): axtname = '-' .join(oglist) with open (f'{name} ' , 'w' ) as axt: axt.write(f"{axtname} \n" ) for spe in oglist: axt.write(fasta[spe] + '\n' ) def sliding (fasta, oglist, sli_len, gap_len, dire ): fa = readfa(fasta) fa_len = len (list (fa.values())[0 ]) posi = 1 while (posi-1 )*gap_len + sli_len <= fa_len-1 : os.mkdir(f"{dire} /posi_{posi} " ) ini_fa = {} sta_po = (posi-1 )*gap_len end_po = sta_po + sli_len for spe in oglist: ini_fa[spe] = fa[spe][sta_po:end_po] fa2axt(oglist, ini_fa, f"{dire} /posi_{posi} /sliding_{posi} .axt" ) posi += 1 if (posi-1 )*3 + sli_len > fa_len-1 : os.mkdir(f"{dire} /posi_{posi} " ) ini_fa = {} sta_po = (posi-1 )*gap_len end_po = sta_po + sli_len for spe in oglist: ini_fa[spe] = fa[spe][sta_po:] fa2axt(oglist, ini_fa, f"{dire} /posi_{posi} /sliding_{posi} .axt" ) for oglist in ogall: ogname = '-' .join(oglist) outputdir = f'SWworkdir_{time_start} /{ogname} ' os.mkdir(outputdir) for gene in os.listdir(fa_dir): genename = gene.split('.' )[0 ] os.mkdir(f'{outputdir} /{genename} ' ) sliding(f'{fa_dir} /{gene} ' , oglist, sli_len, gap_len, f'{outputdir} /{genename} ' ) print ("Sliding window completed...\nRunning Kaks_cal..." ) os.system(f'for i in `ls SWworkdir_{time_start} /*/*/*/*axt`;do echo "KaKs -i $i -o $i.kaks -c {code_model} " >> sliding_window.command; done' ) os.system(f'cat sliding_window.command | parallel --no-notice -j {cpu_num} ' ) os.system('rm sliding_window.command' ) print ("Kaks_cal done\nMerging result..." )def readkaks (kaksfile ): with open (kaksfile, 'r' ) as kf: for line in kf: if line.startswith("Sequence" ): continue dN = line.split('\t' )[2 ] dS = line.split('\t' )[3 ] omega = line.split('\t' )[4 ] return {"dN" : dN, "dS" : dS, "omega" : omega} for oglist in ogall: ogname = '-' .join(oglist) outputdir = f'SWworkdir_{time_start} /{ogname} ' sf_res = open (f'{outputdir} /sliding_window_{ogname} .res' , 'w' ) sf_res.write('Gene\tPosition\tType\tvalue\n' ) for gene in os.listdir(outputdir): genesf = f'{outputdir} /{gene} ' if os.path.isdir(genesf): total_num = len (os.listdir(genesf)) for i in range (1 , total_num + 1 ): kaksinfo = readkaks(f'{genesf} /posi_{i} /sliding_{i} .axt.kaks' ) for v in kaksinfo: sf_res.write(f'{gene} \t{i} \t{v} \t{kaksinfo[v]} \n' ) else : continue print ("Finished." )
运行前需要保证 KaKs 所在路径已经放置在环境变量中。
此后进行计算:
1 $ python sliding_window.py ogfile fa_dir sli_len gap_len cpu_num code_model
ogfile 为一个制表符分隔的文本文件,每一行为一个滑动窗口要计算的物种对。例:
该情况会计算 tax1-tax2 和 tax3-tax4 的滑动窗口结果。
fa_dir 为比对好的 fasta 文件所在文件夹路径。
sli_len 为滑动窗口的长度。gap_len 为每个滑动窗口的间隔。
cpu_num 为使用 KaKs 计算时调用的内核数。code_model 为计算时使用的密码子翻译模式。
运行例:
1 $ python sliding_window.py ogfile fa_dir 57 6 20 1
该情况下滑动窗口大小为 57 bp,间隔为 6 bp,使用 20 个内核计算,并使用标准密码子表。
计算结束后,会在当前所在路径 中,出现一个工作文件夹,每个物种对的结果储存在相应文件夹中以 .res 结尾的文件里。
该文件可用于后续 R 进行可视化。
可视化 使用 R 进行,本来这一步我也想用脚本一步到位地解决,但发现最后要调整的东西很多,所以贴出参考代码然后依照需求修改可能会更好些。
R 代码大致如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 library( ggplot2) library( ggpubr) library( ggprism) library( readxl) barplot = read.delim( "xxx.res" ) color = rep ( "black" , times = length ( barplot$ Type) ) color[ barplot$ value > 1 & barplot$ Type == "omega" ] = "red" barplot = cbind( barplot, color) pdf( "slwd.pdf" , width = 10 , height = 8 ) ggplot( barplot, aes( Position, value, fill = color) ) + geom_bar( stat = "identity" , width = 0.9 ) + theme_prism( ) + theme( axis.line.y= element_line( linetype= 1 , color= "black" , size= 1 ) , axis.line.x= element_line( linetype= 1 , color= "black" , size= 1 ) , axis.ticks.x= element_line( color= "black" , size= 1 , lineend = 1 ) , axis.ticks.y= element_line( color= "black" , size= 1 , lineend = 1 ) , axis.text.x = element_text( angle = 90 , size = 12 , face = "plain" ) , axis.text.y = element_text( size = 12 , face = "plain" ) , axis.title= element_text( size= 18 , face= "plain" ) , legend.position = "none" ) + xlab( "Sliding windows starting positions (bp)" ) + facet_grid( Type ~ Gene, scales= "free" , space= "free_x" ) + scale_x_continuous( breaks = seq( 0 , 300 , by = 100 ) , labels = c ( "0" , "600" , "1200" , "1800" ) ) + scale_fill_manual( values= c ( "#666666" , "red" ) ) dev.off( )
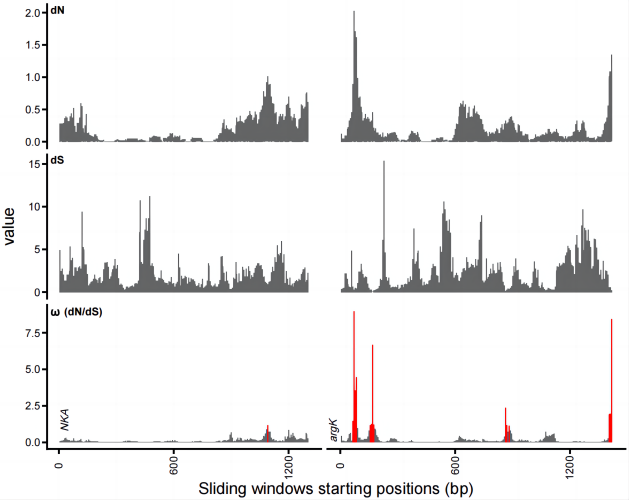
输出图片文件的宽高需根据实际情况修改,随后再调整一下各个标签的位置并给基因名打上斜体,最后的结果就出来了:
其中,ω 标红的区域代表该窗口中 ω 大于 1。
题外话 从作用上来讲,画滑动窗口图,可以了解一个特定的基因内不同区域的选择情况。
从博客经营角度出发,这个又让我能写一篇文,补足前几个月摸鱼的空缺。