比较转录组分析(七)—— 系统发育分析
关于分类学和系统发育分析
分类学,这个学科虽然听起来很简单,但从整个生物史的尺度出发都能算的上是一个非常具有深度且一直以来都争议颇多的学问。从几个世纪前,分类学家就已经致力于通过各种形态学差异或者生活史差异来描述不同物种之间的亲缘关系并且把它们分门别类成不同的类群。在那个时候,不同的学者也会根据 “个人之见” 进行颇具主观性的划分(而且某些近缘物种之间的形态学差异确实可以迥然不同),因此许多分类单元并不统一且一直以来都在不停变动,这一些现象在达尔文先生的《物种起源》中表现得淋漓尽致。
即使古早的分类方法并不完全准确,分类学家依然完成了非常瞩目的成就,就拿现在的物种分类情况来说,很多框架依然与以前一致,并且新的分类单元也是基于已有的框架延申出来的。
测序手段的发展算得上是分类学的一个里程碑,从分子层面的观察让我们对于物种的分类关系有了更为准确的把握,这催生了大量分类错误的纠正以及新物种的发现,并且由此诞生出来了分子系统发育学这一学科。目前很多生信软件都能帮助我们探讨不同物种间的系统发育关系。
这篇文将分享如何通过一些软件来进行系统发育分析。
出现的一些词
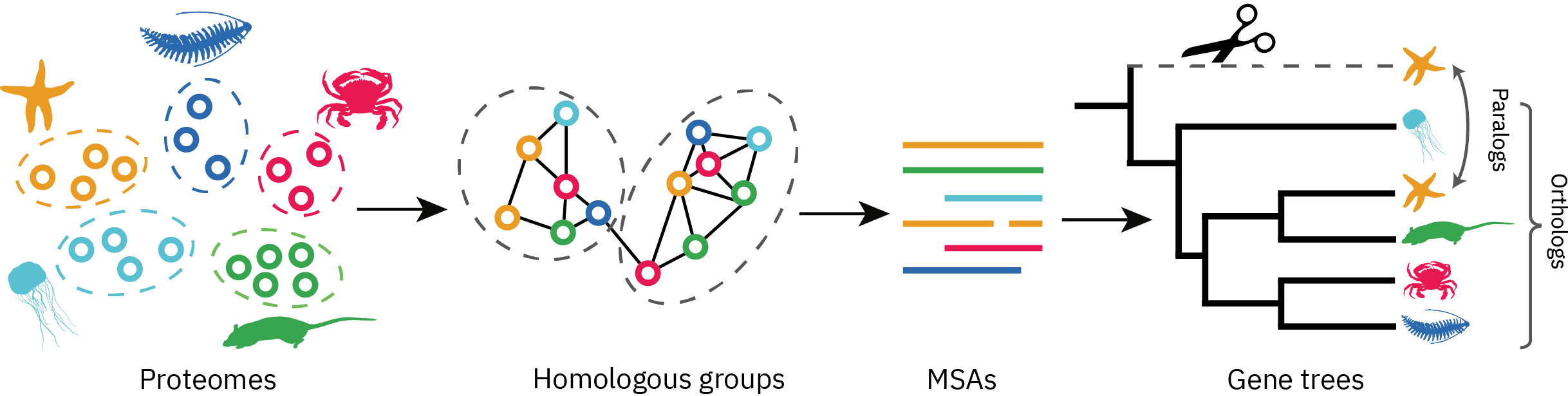
直系同源物和旁系同源物:见 生信基因功能分析工具:Orthofinder 使用教程,更多详细信息见文章末尾题外话知识点。
异同源(xenolog):指通过基因水平转移,来源于共生或病毒侵染所产生的相似基因。也是一个进化学上比较热门的问题,在微生物、病毒及与这些类群密切相关的生物(如昆虫等)中较为常见。
更新日志
2023.06.24 解封文章,完善了关于 Phylopypruner 的说明,补充了各种信息。
2023.07.16 删去了部分内容,进行了一些补充。
2023.09.02 优化了脚本,所需要的时间大大降低了。
分析过程
所需文件和软件
- 不同物种的 pep 序列。
- 软件(相关的安装请自主查阅):
- orthofinder
- mafft
- trimAl
- iqtree (建议使用 v2 以上的版本,模型寻找更快)
- phylopypruner
使用 Orthofinder 寻找同源序列
其他可选的软件也有 OrthoMCL 等,不过从 Orthofinder 具有更突出的优势(见后记题外话), 此外它涵盖了更多且更全面的功能,使用非常便捷,并不需要花过多的时间学习。
安装方法和使用教程见:「基因组学」使用 OrthoFinder 进行直系同源基因分析。
1 | orthofinder -f ./ -t 16 |
这里讲一些需要注意的点:
- 最好指定合适的线程数(因为 orthofinder 默认使用的线程数是 128,不足 128 则使用最大的线程数)。
- 也可以输入 DNA 序列,需要使用参数
-d。 - 无需指定
-M msa,这些工作可以自己做并且更具可控性。 - Orhofinder 在找完同源群后还会为每个同源群进行建树,这会消耗更多的时间,而且它并不会计算自展值,因此如果想省时间在最开始运行命令时指定
-og。 - 跑前请一定确保所有的 pep 序列都已经打上了物种标识!不然在后续的分析中你将很难区分每条序列来自哪个物种。
其他的参数使用默认值即可。理解输出的文件比运行软件更重要,因此这里着重分享 Orthofinder 得到的结果是什么以及有什么用:
有最详尽解释的 orthofinder 官网 https://github.com/davidemms/OrthoFinder。
Comparative_Genomics_Statistics
这个文件夹对于基于转录组分析的结果来说应该是没太大用处的。
它里面包括了一些基因复制事件的信息以及同源群的相关信息(比如说同源群的共享情况、同源群的大小信息等)。
Gene_Duplication_Events 同理,不过里面记录着更详细的基因复制事件信息。
Gene_Trees
里面是关于同源群所对应的基因树。
Orthogroups & Orthologues
更详尽的同源群信息,比如每个同源群中包括了哪些物种的哪些基因。
Orthogroup_Sequences
重要的文件夹,里面包括了所有同源群对应的序列信息,也就是每个 orthogroup 的 fasta 文件。
Single_Copy_Orthologue_Sequences
在同源群中,每个物种只有一个基因的 orthogroup 序列,并不是严格意义上的单拷贝基因(而是推定得到的),如果数量足够可用于序列串联和建树。
Species_Tree
为一份 orthofinder 推断的物种树,默认的方法为 STAG,原理是把包含了所有物种的 orthogroup 对应基因树挑出来,并计算两物种间最近的距离,最后根据每个 orthogroup 的距离矩阵计算得到最后的共识物种树,节点的支持率由这 orthogroup 中的真实情况进行统计计算。
另外一些文件涉及到异同源、HOG 等信息,详细可以前往官网查看。
直接使用 Single_Copy_Orthologue_Sequences 中的序列(情况 ①)
这种方法适用于 orthofinder 找到了很多 ‘single copy ortholog’ 的情况,不过这种一般只在分析的物种数较少时发生。
对 Orthogroup(OG) 进行挑选分类(情况 ②)
进行分类的原因很简单,一些很大的 orthogroup 里肯定包含了很多 paralog,虽然网上对于 orthogroup 的翻译是直系同源群,但显然里面并不都是 ortholog。很多 Phylotranscriptomics 文章中,研究者也会首先挑选出那些较小且包含物种数较多的 orthogroup 进行后续分析。
此外,部分 OG 中包含的物种数量很少,这种 OG 我们也应当剔除分析之外,具体的挑选通过 Python 实现:
1 | # -*- coding: utf-8 -*- |
如果物种标识以下划线区分则将脚本中的 @ 改为 _,用法例:
1 | mkdir ortho_seq |
其中:
-l指定最少物种数-b为当有物种序列数大于多少时该 OG 会被视为大 OG-f为 orthofinder 跑出来的 Orthogroup_Sequences 路径-o为输出的文件夹
上述用例中,该命令将得到叫做 ortho_seq 的文件夹,其中包含 orthogroup_big 和 orthogroup_small 两个文件夹。
之后的操作将在 orthogroup_small 中进行(如果该文件夹处理后依然没得到足够的 msa 则需要对 orthogroup_big 中的序列再跑一次 orthofinder 以进行补充)。
比对以及修剪
在对应的文件夹中输入以下命令:
1 | for i in *.fa; |
1 | for i in *mafft.fasta; |
mafft:
mafft 的 --thread 对应使用的线程数。
此外 mafft 有多种比对模式以适用于不同的序列情况,--auto 开启后则是让 mafft 根据序列情况自动寻找适合的比对策略。
trimal:
trimal 也有很多种修剪策略,不同的修剪策略会选择不同的修剪阈值,目前的 trimal 主要包括三种策略 gappyout、strict 以及 *strictplus*。
第一种只考虑到比对的 gap 部分,会修剪掉比对中的 most gappy fraction。
后两种相较于第一种还考虑到了 gap 部分的相似性分数,其中 strictplus 比 strict 更加严格。
另外还有一种 automated 的方法,它会考虑到一些比对的信息比如说序列数量、序列间的一致性分数等然后基于此选择使用 gappyout or strictplus。
二次筛选
在比对及修剪后部分 msa 的序列长度变短(可能修剪掉太多也有可能是 gap 太多),这些 msa 包含的信息量比较少,因此可以把它们筛掉选择其他那些 “合格” 的 msa 进行下一步的分析。
筛选的思路:选出长度高于一定阈值的 msa,其次删掉里面 gap 过多的序列,然后再根据删除后的物种数情况选择是否保留该 msa。
Python 实现:
1 | # -*- coding: utf-8 -*- |
如果物种标识以下划线区分则将脚本中的 @ 改为 _,用法例:
把修剪后的序列复制一份到新的文件夹中:
1 | mkdir orthogroup_trim |
1 | python /pathto/trim_filter.py -t 30 |
具体的参数含义可见脚本。
在这之后如果一开始使用的是 Single_Copy_Orthologue_Sequences 的序列,那么可以直接跳到序列串联一部分。
构建基因树(情况 ②)
1 | for i in `ls *.trimal.fasta`; |
1 | cat iqtree_command | parallel --no-notice -j 10 |
并行命令使用 IQTREE 进行批量的基因树构建,请根据实际情况修改 -nt 及 -j 参数。
也可以先从小的 Orthogroup sequence 开始建树(更快):
1 | for i in `ls -r *.trimal.fasta`; |
运行 Phylopypruner(情况 ②)
Phylopypruner 是实现多对多到一对一的关键步骤,它的原理是根据基因树所提供的信息进行一系列过滤修剪从而得到最准确的一对一直系同源物(虽然这仍是推定 [putative] 的)。详尽的原理以及各个参数含义可见其 gitlab。
首先需要一个前置脚本 OTU_replace.py 用于替换基因树中的序列 id(iqtree 进行建树时会将 @ 替换成 _,但 Phylopypruner 只识别 @,当然也可以改源码让 Phylopypruner 可以识别 _)。
1 | # -*- coding: utf-8 -*- |
species_list 文件中应当包含所有物种的对应物种标识:
例如如果 fasta 中的序列名为这样,则 species_list 应为其下的格式,其中一行对应一个物种名。
1 | >Human@xxxxxx |
1 | Human |
此后使用 Phylopypruner 推定不同物种的一对一同源物。如果运行所用的数据集过大,在运行的过程中极有可能发生递归错误,因此可以考虑在 Phylopypruner 的 __main__.py 中设置更大的递归深度:
1 | phylopypruner --dir ./ --min-len 100 --trim-lb 5 --threads 6 \ |
上述各参数设置是直接照搬 Phylopypruner gitlab 官网所给的过滤标准最严格的示例参数,但是实际的参数使用应该视自身需求及已有数据集情况而定!
PhyloPyPruner, when compared to pre-existing orthology inference programs, have many more parameters that you could potentially tweak so finding a good starting point might be difficult. These are some suggested settings to get you started, but don't forget to adjust the settings according to your own needs and the overall structure of the dataset that you are working with.
这里附上 Phylopypruner 各参数含义:
--dir多序列比对和树文件所在文件夹。--min-len比对的最小长度,可以根据之前选择的过滤阈值修改。--trim-lb删除长度超过所有分支标准差 n 倍的分支。--threads使用的线程数。--min-support将支持率低于 n 的节点归并成多分枝。--prune使用 xx 方法进行剪枝。--min-taxa不输出物种数小于 n 的比对,可以根据之前选择的过滤阈值修改。--min-otu-occupancy如果某个物种的比对出现率在 n 以下,则删除该物种。--min-gene-occupancy删除包含 1-n 以上 gap 的序列。--trim-freq-paralogs&--trim-divergent&--min-pdist:控制旁系同源过滤和直系同源质量的参数。
--trim-freq-paralogs 4will get rid of taxa with more than 4 times the standard deviation of the total number of paralogs divided by the number of alignments in which each taxon is present in
--trim-divergent 1.25will remove all sequences from a taxon on a per-alignment-basis, if the relative distance from the taxons own sequences, when compared to all other sequences, is than 125%
--min-pdist 1e-8will remove any sequence pair which belong to two different species and have a distance that is less than 1e-8
--jackknife逐个删除物种,返回 删除某个物种后将恢复多少序列和比对 的信息。
如果运行终止并返回 RecursionError 错误,那么你可以在 Phylopypruner 根目录下的 phylopypruner/__main__.py 中对 main() 部分进行修改:
1 | sys.setrecursionlimit(100000) # 添加该行,如果还不行就将该值提高为更高的值,例如 200000 |
对于已安装 conda 并使用 pip 安装的情况,该文件将位于 anaconda3/lib/pythonx.x/site-packages/phylopypruner 中。
最后所得到的伪一对一直系同源基因序列就可以在 phylopypruner_output/output_alignments 中查看,其中每个物种最多存在一条序列,需要注意的是,某些 orthogroup 可能会分离得到多个 xxx_pruned_x.fasta:
1 | OGxxxxxxx.trimal_pruned_1.fasta |
串联序列,Iqtree 建树
此处以最大似然法建树软件 IQTREE 为例,当然也可以视自身要求选择其他方法和对应软件。
串联序列这一部分已在之前的文章中提到过,请走:传送门。
串联后运行下面的命令进行建树:
1 | iqtree2 -s concatenation_ortho.fasta \ |
各个参数的含义:
-s是输入的串联序列文件,-spp则对应前者的分区信息,-nt是使用的线程数(可以使用-nt AUTO让 iqtree 自己选择合适的线程数)。-m是 iqtree 的运行模式,选定MFP+MERGE时 iqtree 会自动寻找最佳分区方案和替换模型。-bb是 iqtree 的一种自展值计算方法 ultrafast bootstrap 的重复次数,一般设置 1000 次。- 注意,ultrafast bootstrap 虽然相较于传统的计算方法只要求更低的计算资源,但是仅当支持率超过 95 时才能说明这个分支是可信的。
-quiet开启静默模式,此时 iqtree 不会有任何的屏幕输出。
更多相关的信息请见 iqtree 的 documentation。
建完树以后会得到一系列文件,以下一些文件比较关键:
xxx.treefile:树文件,为多序列联合建树的结果。xxx.best_scheme.nex和xxx.best_model.nex:有具体的分区信息和模型选择。
而这次得到的树文件,我们就可以把它当作一个揭示了物种间系统发育关系的物种树。
其他方法建树
可见其他文章,例:
题外话
提及到的所有脚本都放在了 github 上,要搬去自用的话需要根据自身情况进行一定的修改等。
本文提出的仅是系统发育转录组学中一种可行的方法套路,请谨慎参考。所有相关脚本及代码的具体参数选择都应依据实际需求进行更正。
如果搞不明白脚本怎么运行想省时间,可以试着看一看博客中的 JuseKit 系列,下载软件并根据教程进行操作(当然更建议学一学 Python)。
Orthofinder 相比之 OrthoMCL 优势在哪
简单来说,OrthoMCL 划分同源群时使用的是 reciprocal BLAST best-hit method(RBH),这种方法有个坏处在于它会受到序列长度的干扰。
基于此,Orthofinder 使用了序列长度进行矫正,提高了同源群划分的精度,这种矫正后的方法叫做 reciprocal best normalised hit(RBNH)。
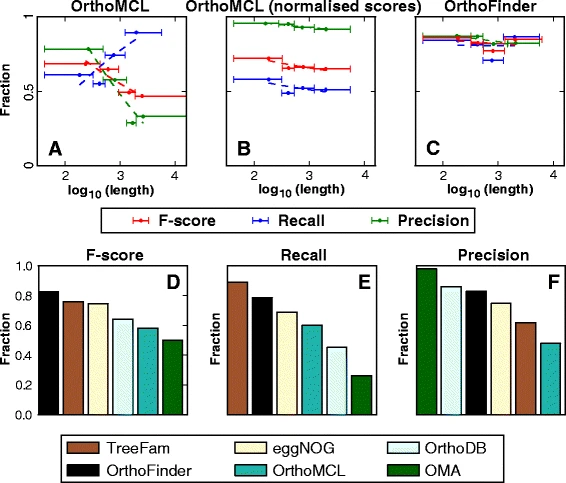
OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy
from Genome Biology
不过这并不代表绝对准确,某些 Orthogroup 中可能仍带有许多的 Paralog。
准确推出 Ortholog 与 Paralog 的必要性
这个问题很简单,以下图为例:
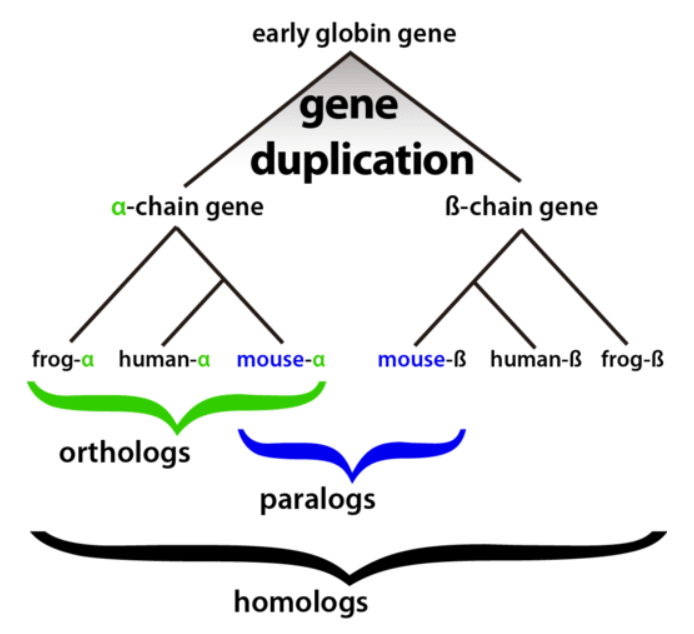
当我们使用直系同源基因进行推断时,不管使用 α 还是 β 都能得到正确的结论,即这三个物种中鼠和人具有更近的亲缘关系。
但如果选错了基因,比如说挑选了青蛙和人的 α 基因,以及鼠的 β 基因,虽然挑取的都为同源基因,但是直系同源和旁系同源的区别会使得我们推断出青蛙和人的亲缘关系更近这一错误结论。
因此,直系同源的正确划分是推断系统发育关系的重点。
蛋白序列建树和核苷酸序列建树
使用核苷酸序列和蛋白序列建树得到的结果大多时候都是一致的,但也有可能存在不同。
原因是核苷酸实际上是一种更 “精” 的序列,它相较蛋白序列而言包含更多的信息,比如说某些同义替换等。
因此当存在出入时一般需要再进行考证,大多数情况下蛋白序列和核苷酸序列建树结果的不一致是出现在某些支持率较低的 node 上,这时再添加一些 Ortholog 分析可能可以解决问题。