
使用 dadi 推断适应度效应(DFE)
前言
适应度效应分布(Distribution of Fitness Effects,DFE)是群体遗传学中的一个核心概念,用于描述新产生突变对生物适应度的影响大小及其分布情况。一般认为,大多数新突变是近中性或有害的,只有极少数能够带来适应性优势。准确推断 DFE,不仅有助于理解自然选择如何塑造遗传多样性,也为遗传负荷评估、疾病变异解释以及进化历史研究提供了重要依据。
在这类分析中,许多基于位点频谱(Site Frequency Spectrum,SFS)的 DFE 推断方法得到了广泛应用。这类方法通常利用中性位点与受选择位点的频谱差异,通过建立群体遗传学模型来估计选择系数的分布。在一个典型的 DFE 推断流程中,通常需要:
- 构建中性位点和目标位点的位点频谱(SFS);
- 根据中性位点估计群体历史(demographic history)参数;
- 在固定人口历史模型的基础上,拟合受选择位点的选择系数分布;
- 根据模型参数推断不同强度有害突变所占的比例,并进一步开展生物学解释。
本文将介绍一个常被用于 DFE 推断的群体遗传学软件 —— dadi。
dadi 使用
GitHub repository: https://github.com/RyanGutenkunst/dadi
官方推荐使用 conda 进行安装:conda install -c conda-forge dadi
提前说明:
- 本文将主要介绍其中 1D demographic DFE 推断的方式(即仅包含一个种群),但阅读本文理解运行原理后,其他的 DFE 推断应该将易于理解。此外,其 GitHub repository 中也有一些详细的 documentation,感兴趣可点击此处跳转进行查阅。
- 在推断 DFE 的过程中,很多数据文件需要自行准备,因此这会对你的生信基础具有一定的要求。考虑到 dadi 的 documentation 中没有相关的指导,本文会尽量详细地对实现过程进行说明,但并不会事无巨细的展开(避免文章篇幅过大)。如果遇到困难,结合 AI 将有助于相关问题解决。
- dadi 的计算核心是种群遗传学中的扩散近似方程,该方程旨在描述种群的等位基因频率分布如何随时间变化,了解这一计算过程也有助于理解其运行背后的逻辑,感兴趣的朋友可以点击上面的文章链接阅读相关的方法部分。
- 下文的代码实操建议新建一个 jupyter notebook 然后逐一同步。
1. 构建中性 SFS,确认种群历史模型,并推断模型参数
dadi 的第一步工作是利用中性 SFS 推断最优的种群历史模型参数。一般来说我们关注的是 CDS 区域的突变,所以一种方法是使用同义变异来构造中性 SFS,这样的好处是同义变异与其他 CDS 内的变异存在连锁,并且经历相近的背景选择,因此在建模时可以捕获这一些效应带来的影响,从而更精确地推断其他编码变异的适应度效应。不过也有一些研究(Huang et al. 2019; Di et al. 2025)会选择使用其他区域的变异:
- Huang et al. 2019:使用了外显子周边的中性区域。该文章认为同义变异本身也受到选择(非中性),这种情况下使用同义变异建模将错误地将选择影响归因到种群历史影响中,导致对非中性区域的适应度效应估计有所低估。
- Di et al. 2025:使用了目标基因组区域附近的中性区域。该文章目的为估计 enhancer/promoter 等基因组元件的突变适应度效应,因此其收集了这些元件附近的中性区域变异进行种群历史模型参数推断。
因此具体的中性 SFS 应当根据自己的研究目的/需求进行确定,此处以同义变异为例,如果你的目的是推断整个 CDS 区域的非同义变异适应度效应,那么你可以:
- 使用 ANNOVAR/VEP 等注释软件,获取所有 coding region 的变异。
- 在这一步中,注释的结果取决于你使用的注释版本,这里的注释将用于后续的一些计算中,可以稍作留意。
- 提取出所有 coding consequence 为 synonymous 的变异,这些变异即是你的中性变异集。
- 提取出所有 coding consequence 非 synonymous 的变异,这些变异即是你的目标非中性变异集(用于后续的 DFE 推断)。
如果你的 VCF 文件里包含个体基因型信息,那么你可以使用 dadi 里自带的函数进行提取:
1 | import dadi |
假设 VCF 文件中的个体全部属于一个群体,你可以使用 bcftools 快速生成一个 pop file 文件:
1 | vcf="xxx" |
如果你的 VCF 文件中不包含个体基因型信息但有等位基因计数信息,你也可以使用以下自定义函数进行提取:
1 | from cyvcf2 import VCF |
注意,这种方式仅适合单群体的情景,如果你想要分析多群体且 INFO 字段里包含不同群体的 AC/AN 值,那么需要进行一定调整才能够使用。
此外,上述简化函数默认每条记录都是双等位变异,并且 AC、AN 均为有效的标量。实际使用前应先过滤多等位位点、缺失 AC/AN 的位点以及不满足质控条件的变异,并且将过滤掉的位点数量从后续的总长度里剔除(见下文),以确保计算的准确性。
提取后,可以看到 dd_neutral 大概格式如下:
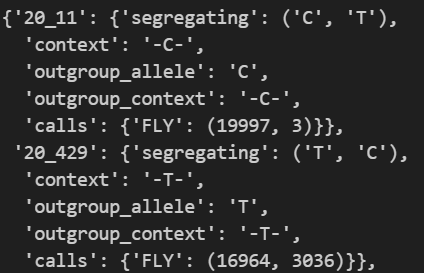
这可以通过 dadi.Spectrum.from_data_dict 进一步转为位点频谱(SFS)。在转换前,需要先确定两个参数:
pop_ids:需要分析的群体名称,必须与 data dictionary 中calls里的群体名称一致。以上文为例,此处即 'POP'。ns:降采样后的染色体数(projection),并不是二倍体个体数。例如,100 个二倍体个体最多对应 200 条常染色体。
降采样可以让不同测序深度、不同缺失率的位点被放到相同样本量下比较。ns 设置过大时,许多 AN < ns 的位点会被丢弃;设置过小又会损失频率分辨率。因此,可以尝试多个候选值,选择能够保留较多位点且仍有足够频率分辨率的 projection。
另一个必须提前确定的问题是 SFS 是否需要极化(polarization):
- 如果能够根据可靠的祖先状态判断 derived allele,可以设置
polarized=True,构建 unfolded SFS。如果设置了极化,则后续参数推断中应该额外加入一个参数以代表祖先等位基因的错判率,且种群模型应使用dadi.Numerics.make_anc_state_misid_func进行包装,这里本文不做拓展,如有需求可点击此处跳转官方 documentation 页面进行深入了解。 - 如果没有可靠的祖先状态,应设置
polarized=False,构建 folded SFS。本文后续将以该点为准进行代码示例分享。
前面自定义函数中的 outgroup_allele 为 -,即没有提供祖先状态,因此这里应使用 folded SFS:
1 | pop_ids = ["POP"] |
需要注意,中性 SFS 和后续的非中性 SFS 必须使用相同的群体、projection 和极化方式。不能用 unfolded 中性 SFS 拟合种群历史,再用 folded 非中性 SFS 推断 DFE。
获得中性 SFS 后,下一步是选择种群历史模型。最简单的模型是假设种群大小恒定:
1 | pts_l = [max(ns) + 120, max(ns) + 130, max(ns) + 140] |
真实群体通常经历过瓶颈、扩张或收缩。这里提供一个稍微更复杂的模型示例,其将历史划分为三个阶段:第一次种群大小变化、第二次种群大小变化,以及从 nu2 指数增长至 nuc 的阶段。
1 | import numpy as np |
在 dadi 中,nu1、nu2 和 nuc 是相对于参考有效群体大小 Nref 的比例;T1、T2 和 Tc 的单位是 2 × Nref 代。因此,这些参数目前都是群体遗传学尺度下的相对量。后续要对相关参数进行相关的推断(如果是恒定大小模型则不用,因为该模型不具有参数),由于数值优化容易停留在局部最优解,所以不应只运行一次。下面对初始值进行多次扰动,并保存每一次拟合结果:
1 | import nlopt |
上面的参数边界和初始值并不是固定模板,需要根据物种的种群历史和预实验结果调整。如果多次优化结果总是落在某个边界上,通常说明边界太窄、模型参数难以识别,或者当前模型并不适合数据。若某个参数需要依据外部证据固定,可以在 dadi.Inference.opt 中通过 fixed_params 指定。
实际分析时,可拟合多个有生物学意义的候选模型,例如恒定种群大小、单次大小变化、瓶颈后增长等,并比较它们的 log-likelihood、AIC 以及残差,确定最适合的群体历史模型。只有基于同一份 SFS 和同一种 likelihood 得到的 AIC 才能直接比较。AIC 可以按下面的方式计算:
1 | k = len(best_params) |
还可以直接检查观测 SFS 与模型 SFS 的拟合情况:
1 | import matplotlib.pyplot as plt |
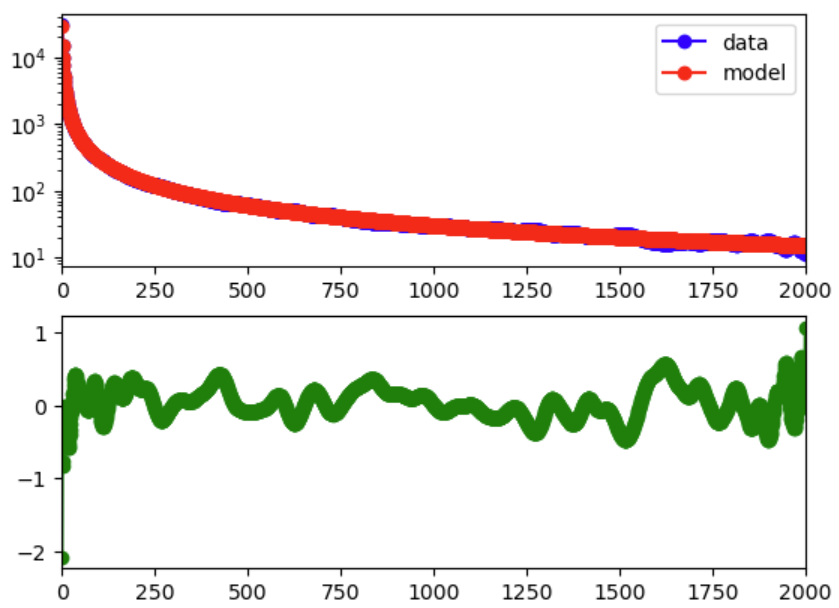
不要只依据更高的 likelihood 选择参数更多的模型。检查结果文件,若模型残差仍然存在明显的频率区间偏差,或者不同重复优化无法收敛到相近结果,则应继续检查数据处理或候选人口历史模型上是否存在问题。
theta0 是由中性 SFS 估计的群体尺度突变率(dadi 推断了一个 SFS 形状,这基础上 theta 相当于丰度)。对于二倍体常染色体:

其中,L_neutral 是用于构建中性 SFS 的可突变位点数,而不是观察到的中性变异数。假设你当前分析的为 CDS 区域,那么你可以先:
- 获取你使用到的注释里 CDS 区域的总长度。
- 将该值减去 uncallable genome 的长度 & 过滤掉的位点数(由于多等位基因),获取实际用于分析的总长度。
- 普遍经验非同义位点的 theta 和同义位点的 theta 比值为 2.5(也有研究认为是 2.31),得 **L_syn = L_total/(1+2.5)**。
以上计算过程应当根据自己的研究需求调整。如果已知这些位点的平均突变率,就可以得到:
1 | L_neutral = 50_000_000 # 根据计算结果调整 |
随后可以把相对种群大小和时间转换为绝对尺度:N1 = nu1 × Nref,N2 = nu2 × Nref,Nc = nuc × Nref,而每个时间参数对应的代数为 2 × Nref × T。这一换算只适用于二倍体常染色体;性染色体、单倍体或其他遗传系统需要使用相应的尺度因子。
2. 生成 DFE cache
DFE 推断需要反复计算不同选择强度下的期望 SFS。如果每一次参数优化都重新求解扩散方程,计算量会非常大。dadi.DFE.Cache1D 会预先计算一系列 gamma 下的 SFS,后续只需按照假设的 DFE 对这些 SFS 加权积分。
这里的选择强度定义为:
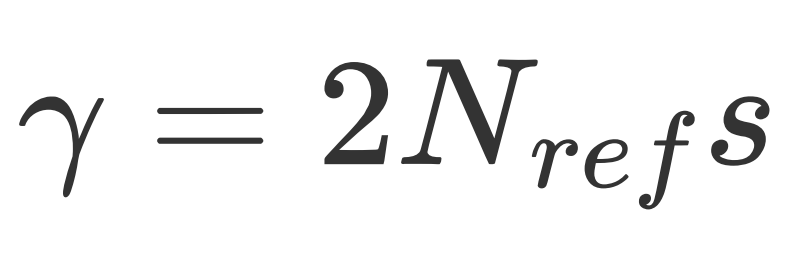
有害突变的 s < 0,因此对应的 gamma < 0。不过,在创建 cache 时,gamma_bounds 填写的是选择强度绝对值的正数范围;Cache1D 内部会将其转换为负值。
生成 cache 时使用的种群历史必须与上一节最终选择的模型完全一致,只需要在参数列表的最后加入 gamma,并在初始化和每一个历史阶段中传入该参数:
1 | import dadi.DFE as DFE |
然后使用上一节的最优种群历史参数生成 cache:
1 | import pickle |
gamma_bounds 应覆盖预期的选择强度范围,gamma_pts 控制该范围内的离散精度,而 pts_l_dfe 控制扩散方程的数值网格。cache 中出现极小的负值可能只是数值误差;如果负值明显小于 -0.001,应增大 pts_l_dfe 后重新计算。cpus 需要根据实际可用计算资源设置,高 CPU 数可有效减少运行时间。
cache 与 projection、种群历史模型及其参数绑定。如果后续修改了 ns、极化方式、种群历史模型或最优人口历史参数,都应重新生成 cache。
3. 构建非中性 SFS,假设选择系数分布,推断选择系数分布参数
非中性 SFS 的构建方法与中性 SFS 基本相同。这里假设已经从注释结果中提取了非同义变异,并生成 selected_vcf:
1 | selected_vcf = "xxx" |
在拟合 DFE 前,还需要计算目标区域对应的 theta_selected。如果中性区域和目标区域的平均突变率不同,应同时校正可突变位点数与突变率:
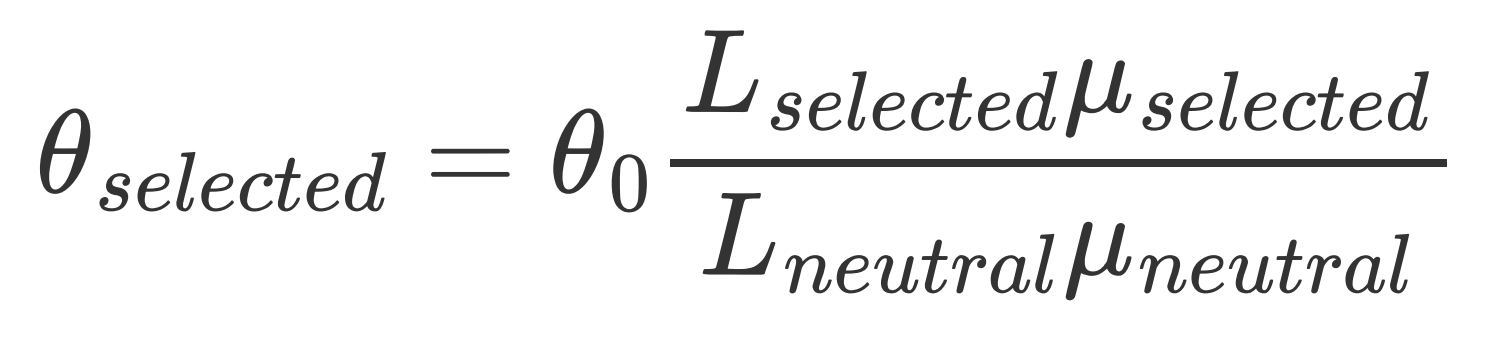
1 | # 对 CDS 区来说可以直接使用 theta_selected = theta0 * 2.31 (或 2.5) |
这里的 L_selected 仍然是可产生目标类型突变的位点数,而不是观察到的非同义变异数。更严格的分析可以按三核苷酸背景统计中性和非中性区域的 mutational opportunities,并结合不同背景的突变率计算有效的 L × mu;亦或者使用已有的 mutation rate map(例如 MuRaL 或 Roulette)进行相关计算。
接下来需要为 DFE 指定概率分布。一个常用假设是有害突变的选择强度绝对值 -gamma 服从 gamma 分布(两个 gamma 的含义不一样,注意区分):

其中,alpha 是 shape,beta 是 scale,平均选择强度绝对值为 alpha × beta。可以直接使用 dadi 内置的 DFE.PDFs.gamma:
这一模型只描述中性至有害突变,不包含 gamma > 0 的有利突变。如果研究对象中有利突变不可忽略,应改用包含有利成分的 mixture DFE,而不是强行用单一 gamma 分布解释全部非中性变异。
1 | sele_dist = DFE.PDFs.gamma |
这里使用 multinom=False,即 Poisson likelihood,因为 theta_selected 已通过中性位点估计,并且目标区域的变异总数包含 DFE 强度信息。如果使用 multinomial likelihood,模型和观测 SFS 都会被归一化,总变异数的信息将被忽略。
拟合完成后,可以生成最优 DFE 对应的期望 SFS,并检查残差:
1 | model_fs_selected = dfe_func( |
alpha 和 beta 描述的是群体尺度参数 -gamma,不能直接当作选择系数 s。对于二倍体常染色体,平均选择系数为:
1 | alpha, beta = best_dfe_params |
如果希望汇报不同选择强度区间中的突变比例,可以将 s 的区间先转换为 -gamma = 2 × Nref × abs(s),再计算 gamma 分布在各区间的概率:
1 | from scipy.stats import gamma as gamma_dist |
到这里你就得到了一个 DFE 推断的结果,后续可以自行编写相关脚本用于可视化各个 selection bin 的 proportion 分布,此处不再做延伸。
作为拓展
- 你可以通过按基因组区块重采样构建 bootstrap SFS,再使用 Godambe information matrix 或重复拟合评估参数不确定性。无论使用哪一种方式,中性和非中性数据都应采用一致的区块划分;同时需要把中性 SFS 带来的
theta0不确定性传播到 DFE 参数中。仅报告单次优化得到的alpha和beta,通常不足以说明结果是否稳定。 - dadi 可以同时考虑多个群体并且将迁移纳入模型计算里,详见其 repository 中的 basic_workflow_2d_demographics.md。
参考资料
Huang et al. 2019: Estimation of allele-specific fitness effects across human protein-coding sequences and implications for disease
Di et al. 2025: The landscape of fitness effects of putatively functional noncoding mutations in humans
Kim et al. 2017: Inference of the Distribution of Selection Coefficients for New Nonsynonymous Mutations Using Large Samples