
使用 PheTK 进行全表型组关联分析
前言
全表型组关联分析(PheWAS,Phenome-wide Association Studies)是一类用于系统性探索遗传变异与多种表型之间关联的研究方法。与常见的全基因组关联分析(GWAS)相比,二者的视角恰好相反:
- GWAS 关注的是“针对一个表型,在基因组范围内寻找哪些变异与之相关”;
- PheWAS 则关注“针对某个特定变异(或一类变异),在众多表型中寻找它会影响哪些性状”。
近年来,随着测序规模的扩大与统计方法的发展,稀有变异(rare variants)在复杂性状中的作用逐渐受到重视。由于单个稀有变异往往难以单独检出显著关联,研究者通常采用 burden analysis 等策略将同一基因中的有害变异聚合起来,以提升统计功效。在典型的 burden 分析流程中,通常会:
- 识别基因中的功能有害变异,例如预测的 loss-of-function 变异或机器学习模型给出的高致病性错义变异;
- 根据样本是否携带这些有害变异进行编码,编码方式可以是简单的二元(0/1),也可以使用携带数量作为计数值;
- 对编码后的变量在多种表型上执行关联分析,从而识别哪些表型与该类有害变异显著相关。
本文将介绍一个基于 Python 实现的 PheWAS 工具 —— PheTK,并展示其在实际分析中的使用方式。
PheTK
文章:PheWAS analysis on large-scale biobank data with PheTK
github repository: https://github.com/nhgritctran/PheTK
安装可以通过 pip 进行:pip install PheTK==0.1.47
目前该软件最新版本为 v0.2.2,但为确保内容无误此处指定的为博主使用的版本,对最新版本感兴趣的朋友也可直接在 github 页面查看详细 guideline。
需要准备的数据
- 样本的 ICD-9 & ICD-10 记录。
- 样本的基本信息(例如性别、年龄、遗传结构前十个 PC 等,用作协变量)。
- 感兴趣的变量(可以是上文提到的稀有变异 burden,也可以是其他个体属性)。
后两者需单独整理成一个 csv 表格,例如:
转换得到样本的 Phecode
由于 ICD-9 & ICD-10 的种类众多,且部分 ICD 指代的疾病症状相近或重叠,因此一种名为 Phecode 的概念被提出,它可以当作是一个粗粒度更大的类别,即一个 Phecode 可能包含多个 ICD code,以更好地概括某一表型,并提高检测关联的效力。
在安装 PheTK 后,可以按照以下流程将样本的 ICD-9 & ICD-10 记录转换为 Phecode 携带信息:
- 首先,根据样本的 ICD-9 & ICD-10 记录,转换为以下格式的表格(csv 格式):
| person_id | flag | ICD |
|---|---|---|
| sample1 | 10 | D17.1 |
| sample1 | 10 | F40.2 |
| sample1 | 10 | Z96.1 |
| sample2 | 10 | F32.9 |
| sample2 | 10 | S30.1 |
其中:
- 第一列
person_id为样本的 ID,需要与后续进行关联分析时使用的样本 ID 对应。 - 第二列
flag对应 ICD 版本,10 对应 ICD-10,如果为 ICD-9 则在对应位置填写 9。 - 第三列
ICD对应具体的 ICD code。
注意,同一个样本可以有多个重复的 ICD 记录(表示 ta 具有该疾病的多次诊断历史)。
- 通过以下 Python 命令将其转换为 Phecode count:
1 | from PheTK.Phecode import Phecode |
此处可调节的参数为 phecode.count_phecode() 中的 phecode_version & icd_version:
phecode_version:指定使用的 Phecode 版本,默认为X,另外的可选项是1.2。icd_version:指定使用的 ICD 版本,默认为US(对应 ICD-10-CM),另外的可选项是WHO(对应 ICD-10)。需根据自身所用数据进行调整,例如此处的场景是使用 UKB 数据,因此使用WHO以对应数据库所采用版本。- 如果有自定义的 Phecode 映射关系,也可以调整参数并使用
phecode_map_file_path指定映射文件,感兴趣的朋友自行探索,此处不做深入介绍。
输出的文件示例(上述命令的 ukb_phecode_counts.icd9_icd10.csv):
| person_id | phecode | count |
|---|---|---|
| samplex | ID_089 | 2 |
| samplex | GU_602 | 1 |
| sampley | BI_160 | 1 |
| samplez | GI_522.9 | 1 |
| samplez | GU_582.2 | 1 |
输出的 Phecode count 文件中:
- 第一列
person_id与步骤 1 中的文件相同 - 第二列
phecode对应 Phecode 体系中的编码,第三列count表示该个体具有多少该 Phecode 的记录。
如果你对 ICD code 和 Phecode 之间的映射关系感兴趣,可以在 jupyter notebook 中运行以下命令查看:
1 | from PheTK import _utils |
进行 PheWAS
在处理完上述数据后,使用 PheTK 即可快速进行 PheWAS:
1 | python -m PheTK.PheWAS \ |
各个参数的含义:
--phecode_version:使用的 phecode 版本,应与phecode.count_phecode()使用的一致。--cohort_csv_path:可见前文部分 “需要准备的数据” 中的说明,该 csv 文件的列中需要包含--covariates和--independent_variable_of_interest指定的变量,此外需要有列名为 "person_id" 的列对应样本 ID。--phecode_count_csv_path:输出的 Phecode count 文件。--sex_at_birth_col:性别信息对应的列名,在某些性别特异的 Phecode 里需要根据性别信息取出特定性别的个体(例如流产只有女性、精子畸形只有男性)。--male_as_one:性别列中,男性的编码方式是否为 1,如果是则填写True。--covariates:拟合模型时使用的协变量。--independent_variable_of_interest:感兴趣的目标变量。--min_case&--min_phecode_count:前者用于筛选 case(可一定程度上筛选掉偶然患病的情况,但也可能错失真实信号),后者用于过滤包含病例过少的 Phecode。此处即:当 phecode count 至少为 1 时,才将该个体视作病例,并过滤包含病例不多于 50 个的 Phecode。--threads:使用的线程数。需注意部分 Scipy 版本下该参数的调节不起作用,建议配合 slurm 等作业调度系统对其使用的线程数进行限制,过高的线程数有时并不会加快运行速度。--output_file_name:输出的 PheWAS 结果,可基于该结果进行可视化。
可视化也可以通过其内置的绘图函数进行:
1 | import PheTK.Plot |
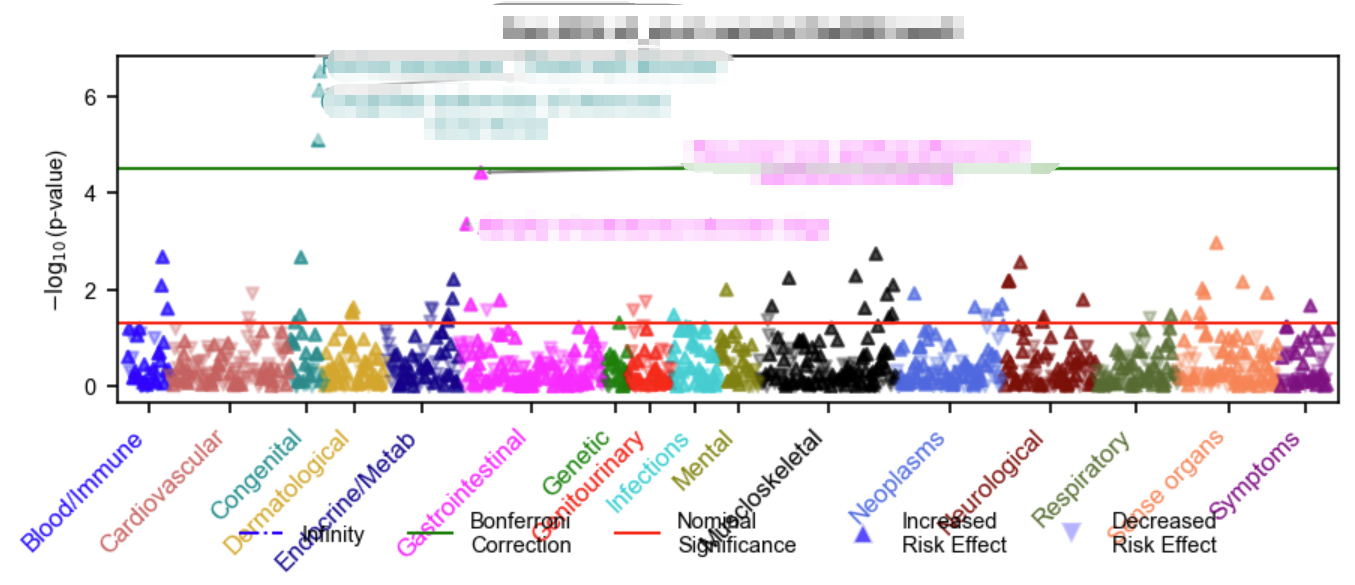
补充
本文章介绍的 PheTK 主要是根据个体的 ICD 携带情况,对其做分类后,再将每一种 Phecode 视作一种表型进行关联分析。
其主要的原理即构建逻辑斯蒂回归模型,并进行拟合得到相关的统计量。因此,如果你关注的全表型组和本文所说的 ICD 有所区别,你也可以按照这种思路自行进行相关的 PheWAS。事实上,你仅需将你关注的表型组整合到上述所说的 cohort_csv 中,你就可以自行构建模型并对每个表型进行相关的关联测试(例如这篇文章)。
作为参考,PheTK 进行关联分析的方式你可以详细浏览其源码中的 phewas.py (link)。如果你关注的表型是连续型数值而非二元分类,你可以考虑将相关的模型替换为最小二乘回归(sm.OLS)。
声明
本文章的前言部分使用了 ChatGPT v5.1 进行润色,并人工核对了润色后内容的准确性。