
使用 igv-reports 进行基因组数据可视化
基因组数据分析中,Integrative Genomics Viewer(IGV)以其直观的交互式界面和丰富的可视化功能,成为我们查看变异和注释等基因组信息的首选工具。然而,在某些情景下 IGV 的可视化优势难以发挥:基因组数据在服务器上,但在服务器中无法方便地调用图形页面,而基因组数据下载到本地再使用 IGV 可视化又过于不便且耗时(特别是在数据量庞大的情况下)。
针对这一问题,igv-reports 成为了一个很好的替代工具。它能够将 IGV 的会话内容和基因组浏览快照一并打包成静态 HTML 报告。生成的报告文件下载到本地后,用户可直接在浏览器中查看和交互,操作体验与 IGV 类似,同时支持灵活分享,兼顾效率与便捷性。
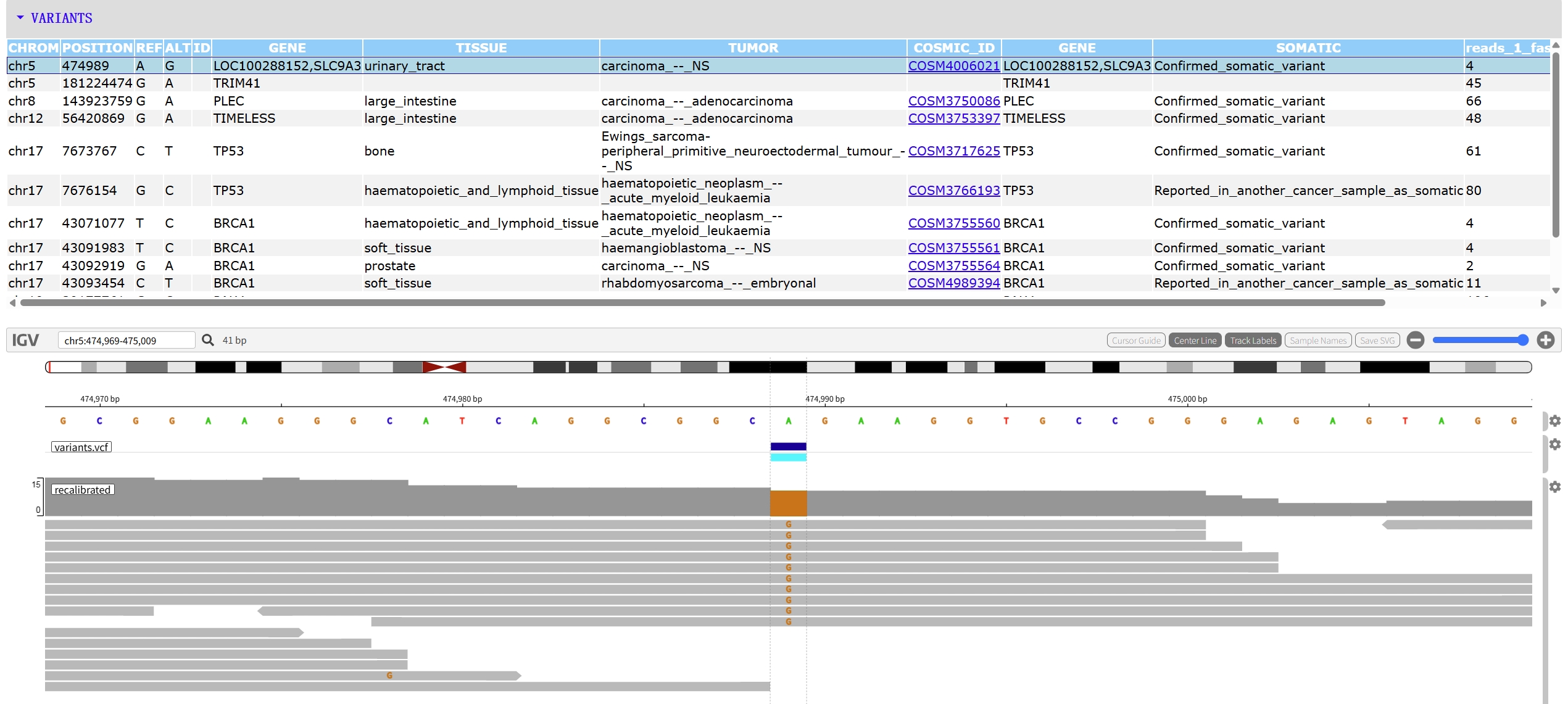
需要注意的是,igv-reports 适用于可视化基因组部分区域时使用,例如你关注的是某些变异位置或特定基因附近的比对情况/信号时,使用 igv-reports 是合适的。但如果你想要大尺度地进行可视化(例如展示整条染色体的变化情况等),该方法可能具有一定局限性(见下文)。请根据自己的需求选择合适的方案。
igv-reports 安装
关于 igv-reports 的运行,最全面的资料请前往 igv-reports github page。
igv-reports 需要 python 3.8 及以上版本,可使用 conda 或 pip 安装:
1 | conda |
如果遇到依赖问题,可检查是否是 pysam 未正常安装,如果是,可通过 conda 解决:
1 | conda install bioconda::pysam |
igv-reports 使用
igv-reports 可用于多种类型的数据可视化,本文将详细介绍其中一个使用示例,涉及到可视化变异位点及其附近的比对情况。为了复现该使用示例,你可以运行以下命令以克隆 igv-reports repository 并移动至有关文件夹:
1 | git clone https://github.com/igvteam/igv-reports.git |
使用示例
完成上述操作后,运行以下命令:
1 | create_report test/data/variants/variants.vcf.gz \ |
输出的页面概览可见(结合该页面以理解下述参数说明):https://igvteam.github.io/igv-reports/examples/example_vcf.html
重点理解这里面各个参数的作用:
- 输入文件为
test/data/variants/variants.vcf.gz,除了 VCF 文件外,也可提供 BED 或 MAF 等以制表符分隔的格式文件。需注意使用 VCF 文件时部分参数的表现会有些许不同。 --genome:指定参考基因组版本。也可使用--fasta替换该参数并自行指定基因组序列文件路径(需建好索引)。--ideogram:染色体带型文件路径,用于绘制染色体带型,非必需。--flanking:指定目标区域两侧扩展的碱基数(默认 1000),仅扩展后的区域中 track 信息会被保留。--info-columns:包含在页面上方表格(此例中即变异表格)中的信息字段名称列表。如果输入的文件为 VCF,则这里给定的列表对应 INFO 列中的字段。如果输入的文件为其他制表符分隔格式,则对应列名。
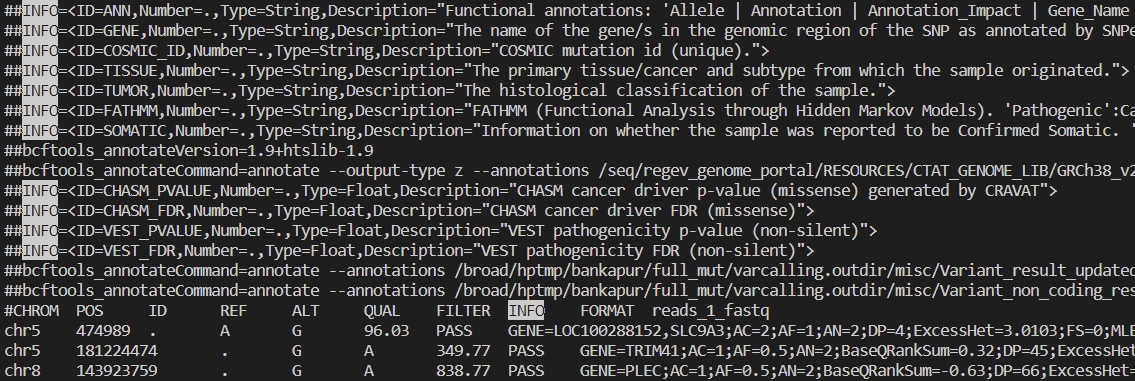
--samples&--sample-columns:包含在变异表格中的样本信息。--samples未指定时默认使用全部样本,--sample-columns指定具体展示样本的哪些信息(此例中即展示深度 DP 及基因型质量 GQ)。--tracks:展示的 track,支持多种文件类型,如比对文件(BAM/CRAM)、变异数据(VCF)、注释文件(GFF3/GTF)及基因模型(UCSC genePred)等。此处分别指定了变异 VCF 文件、比对文件和 UCSC genePred 注释文件。--output:输出的 html 报告文件。
最详细的参数说明可见:https://github.com/igvteam/igv-reports?tab=readme-ov-file#creating-a-report
更多的命令示例可见:https://github.com/igvteam/igv-reports?tab=readme-ov-file#examples
此处将额外提及一些其他需要注意&使用场景更多的参数。
其他场景及需注意参数
使用除 VCF 文件以外的制表符格式文件时:
--sequence参数用于指定染色体名称所在的列数。--begin及--end分别对应起始位置和终止位置。- 使用
--info-columns可以限制展示的信息。
在 track 中包含 BAM/CRAM 等比对文件时:
--exclude-flags用于过滤比对,其默认值为 1536(即过滤重复和不可靠的比对)。将该值设为 0 将不做过滤。
有关局限性
经过测试,igv-reports 的主要时间开销在于加载 track 文件和基因组文件中,因此运行速度上不需要过于担心。但需要注意,其输出文件的大小与展示的区域大小有关,上述示例的输出文件仅 1mb 左右是由于其仅涉及到 13 个变异位点及其两侧 1kbp 区域,如果想可视化整条染色体序列,输出文件就会大很多(作为参考:我使用 5 个 track 的人类 22 号染色体可视化结果文件大小为 1.45G)。
综上,若需展示大范围区域(如整条染色体),建议优先使用 IGV 本地客户端。igv-reports 更适用于靶向分析(如特定基因或变异位点),以平衡可视化效果与文件体积。