
trim_galore 后 Per Base Sequence Content 出现问题的原因
最近在处理基因组数据时,遇到的大多数据都已经处理过,所以 fastQC 里基本没有问题,可以直接用来比对。不过这几天有一个数据比较异常,所以按照惯例地跑了一下 trim_galore,发现了一个意料之外的情况:
- 质控前,所有数据都有 Adapter Content 的警告,但是其他方面没有问题:
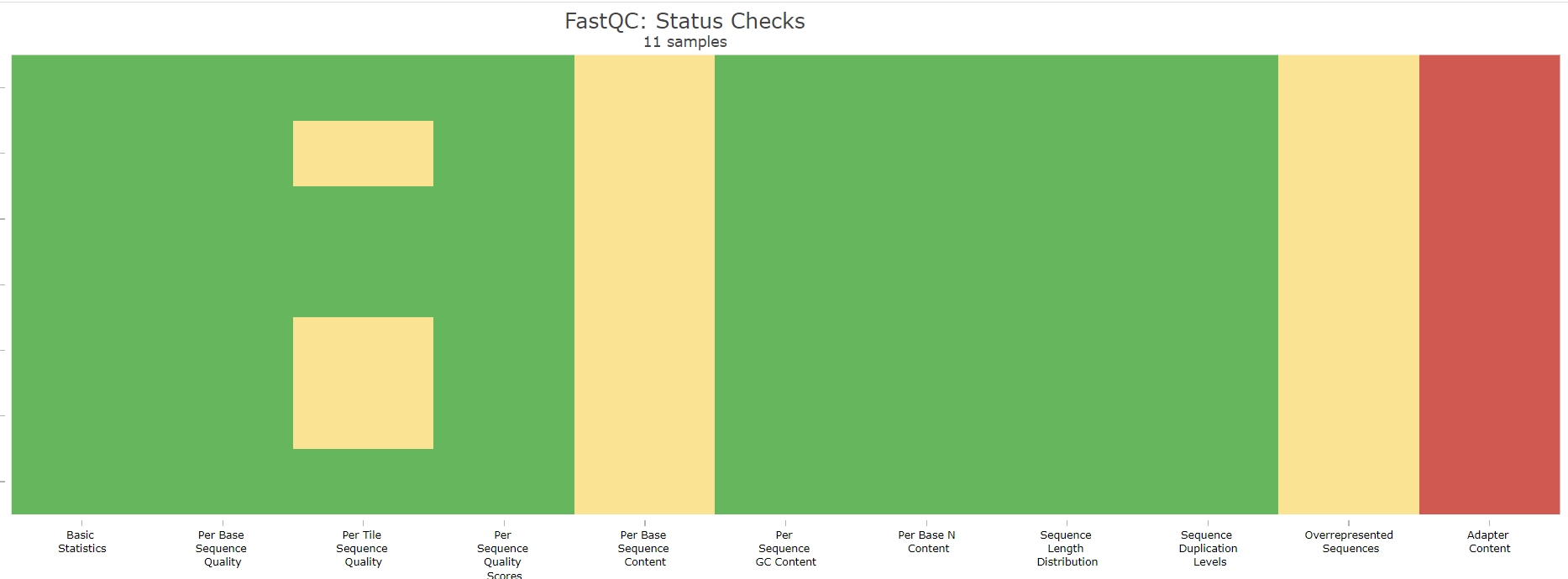
- 质控后,Adapter Content 的警告消失,但是 Per Base Sequence Content 全部爆红:
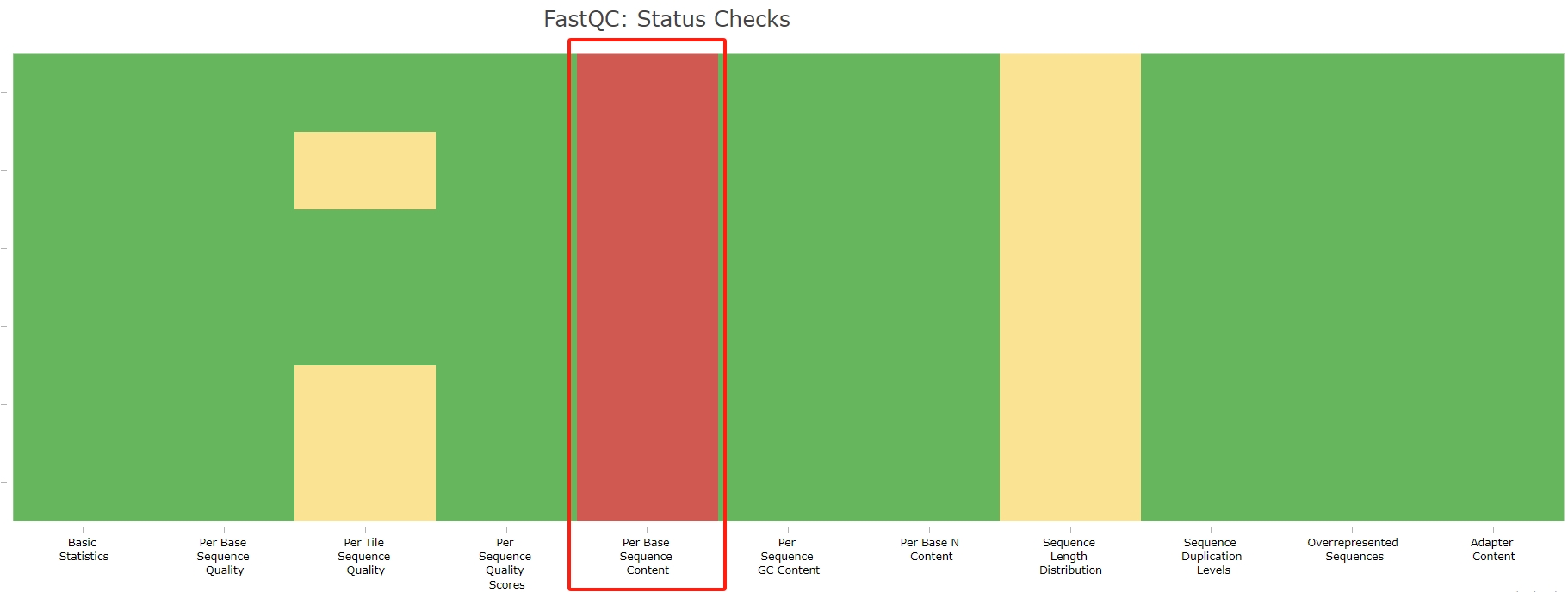
经过比较,原因都是 reads 末端的碱基组成异常导致:
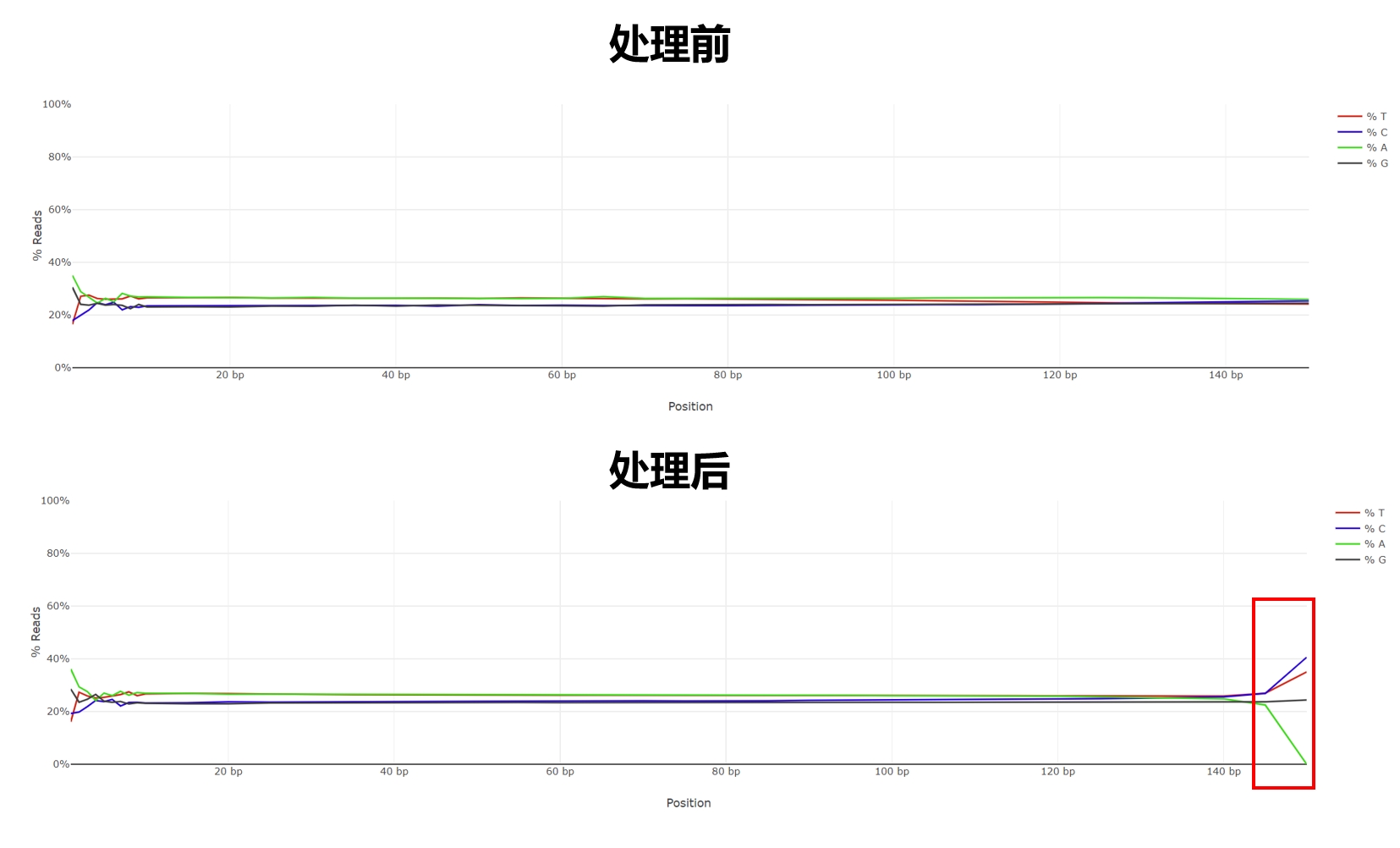
经调查在 Trim Galore github 中已经有人提到过(issue #81),这里简单阐述下原因:
Trim Galore 在过滤时采用非常严格的策略,具体来说,它并不只在接头完全匹配时才进行移除,而是在末端开始进行逐个匹配。例如,假设识别到的接头序列为 AGATCGGAAGAGC,而某一个 reads 从末端开始为 ACCTCG,虽然这里从第二个碱基开始就不能和接头匹配上,但是由于第一个碱基 A 和接头相匹配,因此被 Trim Galore 移除,这也是为什么能在运行 trim_galore 后发现最末端位置上碱基 A 的比例是 0。
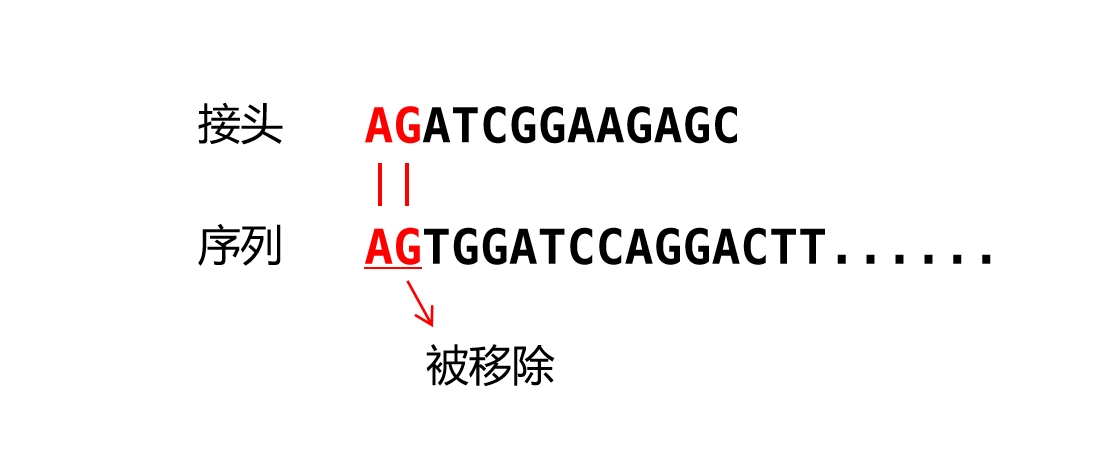
这一行为的背后动机是在亚硫酸氢盐测序数据分析中 ,reads 里如果掺入人工序列将会对结果造成很大影响,因为每个碱基都可能参与最终的甲基化状态判断,所以此处采用了一个看上去略显极端的方式来避免该问题的发生。调整这一行为的方式也很简单,trim_galore 的参数 --stringency 默认指定为 1,将其提升到 n 就会使 Trim Galore 在至少 n 个碱基与接头匹配时进行移除。不过此处开发者强调即使默认设置会使数据在 fastQC 里表现异常,但它大概率不会对之后的 mapping 产生任何影响。
在此记录,希望能帮助到其他有同样疑惑的人。
本博客所有文章除特别声明外,均采用 CC BY-NC-SA 4.0 许可协议。转载请注明来自 Juse's Blog!
评论