******************************************************************** * US-align (Version 20240730) * * Universal Structure Alignment of Proteins and Nucleic Acids * * Reference: C Zhang, M Shine, AM Pyle, Y Zhang. (2022) Nat Methods* * C Zhang, AM Pyle (2022) iScience. * * Please email comments and suggestions to [email protected] * ********************************************************************
******************************************************************** * US-align (Version 20240730) * * Universal Structure Alignment of Proteins and Nucleic Acids * * Reference: C Zhang, M Shine, AM Pyle, Y Zhang. (2022) Nat Methods* * C Zhang, AM Pyle (2022) iScience. * * Please email comments and suggestions to [email protected] * ********************************************************************
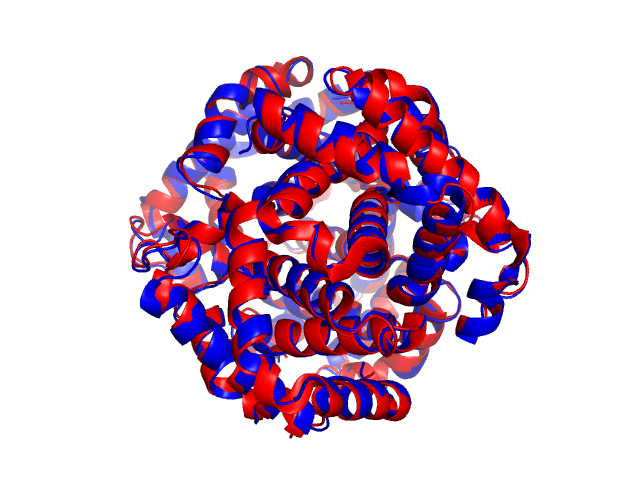
Name of Structure_1: ./2PGH.cif:A:B:C:D (to be superimposed onto Structure_2) Name of Structure_2: ./3GOU.cif:C:D:A:B Length of Structure_1: 574 residues Length of Structure_2: 574 residues
Aligned length= 574, RMSD= 1.03, Seq_ID=n_identical/n_aligned= 0.815 TM-score= 0.98690 (normalized by length of Structure_1: L=574, d0=8.41) TM-score= 0.98690 (normalized by length of Structure_2: L=574, d0=8.41) (You should use TM-score normalized by length of the reference structure)
(":" denotes residue pairs of d < 5.0 Angstrom, "." denotes other aligned residues) VLSAADKANVKAAWGKVGGQAGAHGAEALERMFLGFPTTKTYFPHFNLSHGSDQVKAHGQKVADALTKAVGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHHPDDFNPSVHASLDKFLANVSTVLTSKYR*VHLSAEEKEAVLGLWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSNADAVMGNPKVKAHGKKVLQSFSDGLKHLDNLKGTFAKLSELHCDQLHVDPENFRLLGNVIVVVLARRLGHDFNPDVQAAFQKVVAGVANALAHKYH*VLSAADKANVKAAWGKVGGQAGAHGAEALERMFLGFPTTKTYFPHFNLSHGSDQVKAHGQKVADALTKAVGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHHPDDFNPSVHASLDKFLANVSTVLTSKYR*VHLSAEEKEAVLGLWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSNADAVMGNPKVKAHGKKVLQSFSDGLKHLDNLKGTFAKLSELHCDQLHVDPENFRLLGNVIVVVLARRLGHDFNPDVQAAFQKVVAGVANALAHKYH* ::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::.*:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::.*:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::*:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::.* VLSPADKTNIKSTWDKIGGHAGDYGGEALDRTFQSFPTTKTYFPHFDLSPGSAQVKAHGKKVADALTTAVAHLDDLPGALSALSDLHAYKLRVDPVNFKLLSHCLLVTLACHHPTEFTPAVHASLDKFFAAVSTVLTSKYR*VHLTAEEKSLVSGLWGKVNVDEVGGEALGRLLIVYPWTQRFFDSFGDLSTPDAVMSNAKVKAHGKKVLNSFSDGLKNLDNLKGTFAKLSELHCDKLHVDPENFKLLGNVLVCVLAHHFGKEFTPQVQAAYQKVVAGVANALAHKYH*VLSPADKTNIKSTWDKIGGHAGDYGGEALDRTFQSFPTTKTYFPHFDLSPGSAQVKAHGKKVADALTTAVAHLDDLPGALSALSDLHAYKLRVDPVNFKLLSHCLLVTLACHHPTEFTPAVHASLDKFFAAVSTVLTSKYR*VHLTAEEKSLVSGLWGKVNVDEVGGEALGRLLIVYPWTQRFFDSFGDLSTPDAVMSNAKVKAHGKKVLNSFSDGLKNLDNLKGTFAKLSELHCDKLHVDPENFKLLGNVLVCVLAHHFGKEFTPQVQAAYQKVVAGVANALAHKYH*

{kind=link}