
DNA 语言模型 GPN 在突变效应预测中的应用及表现
众所周知,语言模型已经在诸多领域得到了广泛应用。受深度学习狂潮影响深远的生物学领域里,出色的语言模型应用不断涌出。以 DeepMind 为例,其所开发的 Alphafold、Alphamissense 等都是基于语言模型实现的。
这篇文章将分享一个最近发表的 DNA 语言模型(GPN,Genomic Pre-trained Network)和它的变体 GPN-MSA,该模型与之前的 DNABERT 等 DNA 语言模型不同,其使用单个碱基作为 token 进行训练,并在无监督的突变效应预测中取得了良好的成效。
相关论文链接见文章结尾,本文章将重点介绍两个模型的设计和它们之间的区别,并写出一些很值得学习的地方。
GPN
GPN 是一种自监督训练的 DNA 语言模型,与 GPN-MSA 不同的地方在于,它可以仅依靠未经过比对的数个基因组序列而不使用多序列比对(MSA)进行训练,此外它使用的为单纯的 DNA 序列而未使用到任何功能基因组信息,因此可以说 GPN 的泛用性是非常强大的,因为对于很多类群中的生物来说,获得跨物种的 MSA 和全面的功能基因组数据并不是容易的事情。
GPN 的模型设计如下:
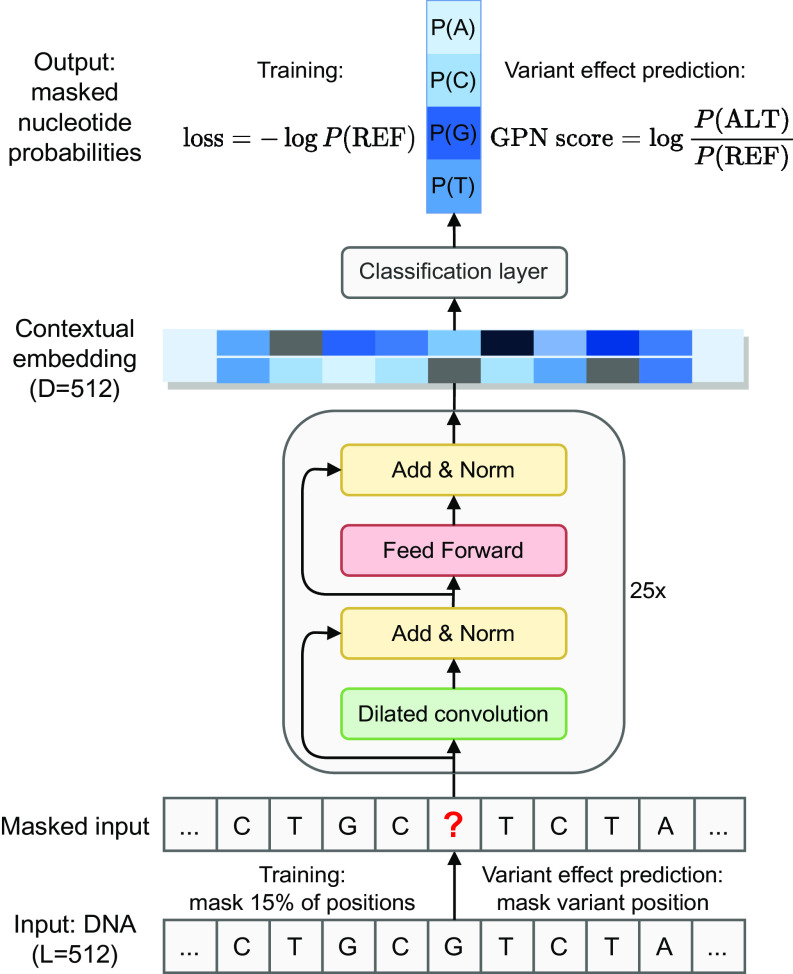
该模型的训练架构类似于 Transformer,其中 DNA 的输入遵守如下规则:
- 在基因组上使用
512bp的窗口 &256bp的步长进行采样(类似于滑动窗口),并使用反向互补序列作为数据增强手段(DNA 语言模型中常见的方法)。 - 该采样并不针对全基因组,而是着重选择了特定区域(如外显子、启动子)以及和这些区域等量的其他随机窗口。
- 对于重复区域进行了损失权重的调整,以改善模型表现。
对于输入的 DNA,其随机掩码 15% 的碱基,并传入给类 Transformer 模块。值得注意的是,与 Transformer 不同,这里用空洞卷积替代了多头注意力机制,文章的理由如下:
- 卷积神经网络比用注意力机制收敛地更快。
- 对于该规模的 DNA 序列而言,卷积神经网络的局部模式识别可能较注意力机制的全局模式识别更加有用。
- 卷积的线性复杂度对于其向更长序列的推广是有益的。
对于最后一点,具体来说,如果我们使用的为卷积神经网络,那么不论是处理 512 碱基还是 1k 个碱基,卷积层对每个碱基的计算成本是固定的。所以,处理后者大概需要的计算量是前者的两倍。
相比之下,注意力机制涉及到序列中所有元素之间的关系,其复杂度是序列长度的平方。在处理长序列时,这会导致计算成本急剧增加。为了处理长序列,模型可能需要将序列分成更小的块,并分别对它们进行处理,然后再将结果整合起来。这种分块处理通常需要在块之间有重叠部分,以捕捉块边界处的上下文信息,这使得整个处理过程变得复杂。

输入的掩码序列经过 25 个类 Transformer 块后被处理成包含上下文信息的 embedding,该 embedding 则被输入到后续的分类层中计算特定位置上四种碱基的概率,模型的损失函数为预测为正确碱基的概率的负 log 值。
此后,Benegas 等人利用该模型进行了一系列的分析工作,所得结果大致如下:
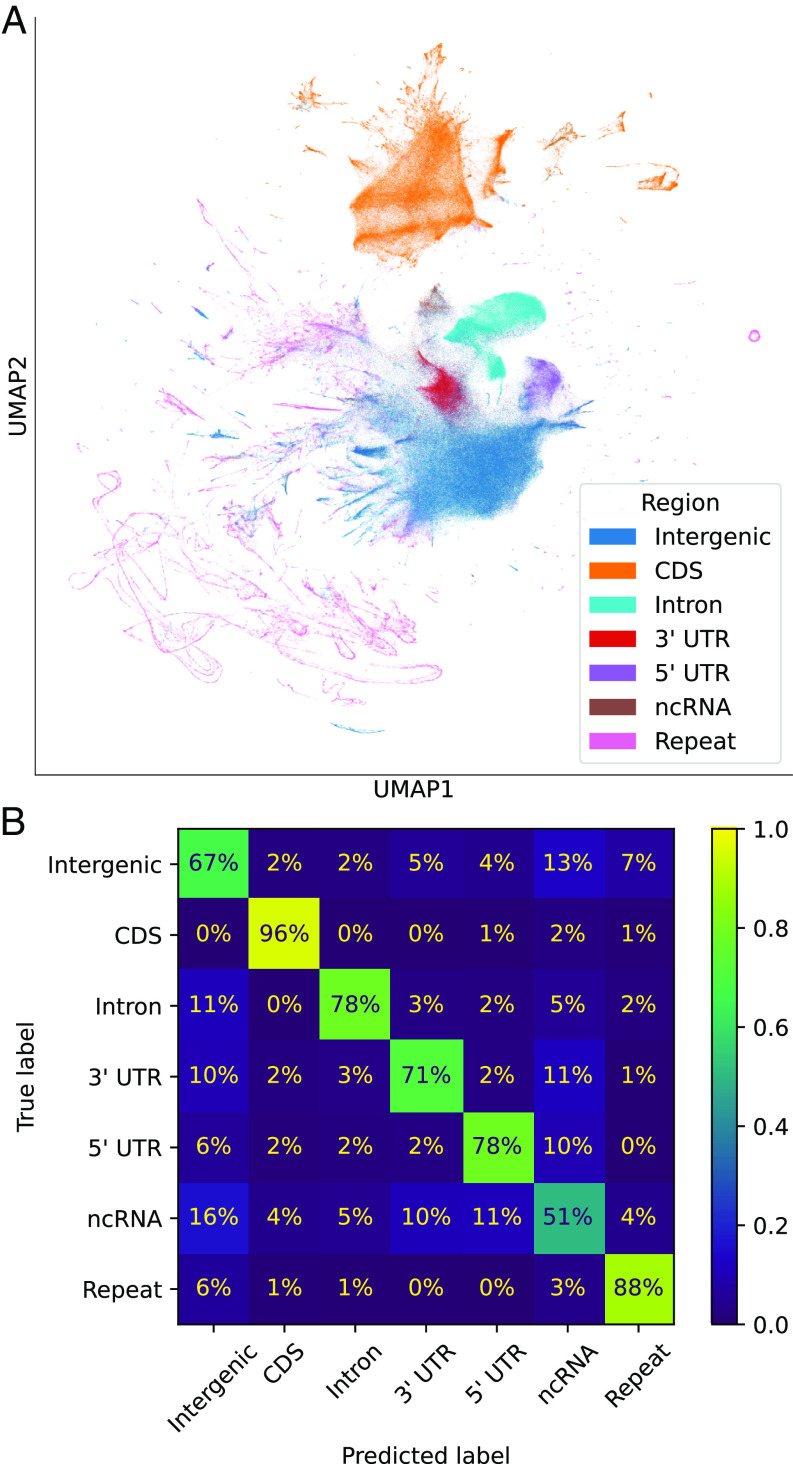
- 在无监督情况下,模型也能有效识别部分基因组区域。使用 embedding 训练逻辑斯蒂回归分类模型后,在 CDS 上实现了高准确率(96%),但对于 ncRNA 和基因间区,模型的表现较为糟糕(51% 和 67%)。
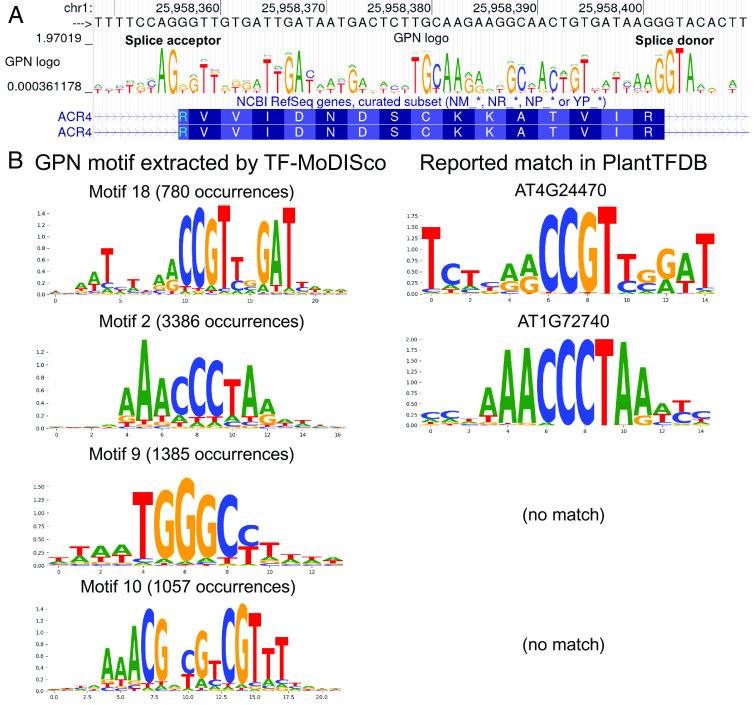
- 通过单独掩盖基因组上各个位置的碱基,模型鉴定出了一些可以在数据库中匹配到的基序和一些尚未匹配但在其他研究中有被提及或者序列特征较特殊的基序。
- 通过以
P(突变碱基)/P(参考碱基)的log值作为突变效应预测的分数,模型实现了良好的预测性能,对于那些因为与 functional variant 连锁的 neutral variants,模型也能实现有效的判别。

可以看出,GPN 的模型设计其实也并不复杂,但这里采取的一些方法令人较为受益。例如,不同于以往的研究,GPN 将注意力机制换为传统的卷积层并在单个基因组上取得了良好的学习效果。此外 GPN 使用了一种巧妙的无监督方法,将预测的不同碱基概率之比巧妙地转换为 variant effect 的 score。虽然这些想法来源自不同的研究,但具体的实现也要研究者巧妙地把它们组合起来。
GPN-MSA
如果说 GPN 是一个轻巧的、适用于多数非模式物种的工具,那么 GPN-MSA 就是专门设计用于人类基因组的语言模型。与 GPN 不同,GPN-MSA 舍弃了卷积层重回正统的 Transformer 架构(RoFormer),同时它的输入数据也由单个基因组变成了九十种脊椎动物的 MSA。
不难看出,在这种情况下,更关注全局信息的 Transformer 会有更好的表现。这也告诉我们,对于不同的数据,灵活修改并使用合适的模型架构是非常重要的。

它的数据处理大致如下:
- MSA 处理,其首先下载了一百种脊椎动物的多序列比对,对其进行一定处理后,去除了与人类亲缘关系最接近的十种灵长类动物。
- 训练输入的数据为长度
128bp步长64bp的窗口,与 GPN 相同,这里有选择性地对窗口进行了选择:①、专注于功能重要的区域,通过保守性分数 PhastCons 选择所有窗口中平均保守性分数最高的 5% 和剩余窗口随机选择 0.1%;②、降低了重复序列的权重并提高保守序列的权重(同样通过 PhastCons 进行)。 - 该模型同样使用反向互补序列作为数据增强手段,同样掩码 15% 的核苷酸进行训练。与 GPN 不同的是,该模型在数据增强中,还将非保守区域的参考碱基随机替换为其他碱基(在计算 loss 时),此外该模型使用 21 号染色体作为早停法的测试 loss 参考,且保留 22 号染色体作为进一步的测试(但在论文中并未使用到)。
去除灵长类动物的原因文章并没有详细提及,但是可能原因也较好理解。首先这些生物与人类的基因组序列相似性可能很高,因此它们可能没法突出那些更广泛进化范围内保守的区域。此外,高度的相似性也意味着数据的冗余信息可能较多,因此去除将有利于模型更有效地提取特征。
另外,文章还进行了消融实验,以确定不同的模型架构对于最终模型表现的影响,具体的尝试可见论文的方法部分,此处贴出原文内容:
- w/o MSA: the model is only trained on the human sequence, without access to other species.
- MSA frequency: variants are scored using the log-likelihood ratio of observed frequencies in the MSA column, with a pseudocount of 1.
- Train on 50% most conserved: expand the training region from the smaller 5% most conserved to a larger set with less overall conservation.
- Include closest primates: do not filter out from the MSA the 10 primates closest to human.
- Don’t upweight conserved: do not upweight the loss function on conserved elements.
- Don’t replace non-conserved: do not replace the reference in non-conserved positions with random nucleotides when computing the loss function.
Modeling the single human sequence instead of the MSA has by far the biggest impact. Using the column-wide MSA frequencies as predictor also shows a large decrease in performance. Including primate species close to human, or training on less conserved regions, have a moderate impact on performance. Finally, of relatively minor impact are removing the upweighting of conserved elements or removing the data augmentation procedure of replacing nucleotides in non-conserved positions.
目前 Biorxiv 论文上研究人员仅将该模型在疾病预测中的表现进行了展示,在突变致病性预测中,GPN-MSA 超越了基于功能基因组数据的 CADD 以及保守性分数 PhyloP,在多个数据集(Clinvar、COSMIC)里取得了 SOTA。

当然,GPN-MSA 在利用多序列比对信息的同时,也失去了其在比对质量较差的非编码区上的表现机会,在文章的最后作者提到了一些未来的方向:
- 整合群体遗传变异信息,而非依靠单个基因组。
- 整合 DNA 序列和功能基因组学信息。
最后,我个人认为这个工作对我的启发还是非常大的,归根结底这个模型设计其实并不算非常复杂,但是很多思路缺一不可。此外这个架构还可以用在多个其他领域的下游分析中,它还有很多利用价值等待我们挖掘……
文章链接如下:
DNA language models are powerful predictors of genome-wide variant effects
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10622914/
GPN-MSA: an alignment-based DNA language model for genome-wide variant effect prediction
https://www.biorxiv.org/content/10.1101/2023.10.10.561776v1.full
