
系统发育转录组学是否靠谱?
前言
我一直认为,阅读文献应该是带着明确的目的去读,要么是想要从中学习到什么东西,要么是想通过它解答一些问题。
作为开篇,我先来分享一下对我正在研究的东西有支持性的文章:
Is Phylotranscriptomics as Reliable as Phylogenomics?
from Mol Biol Evol
事实上,很久之前我认为用转录组找出来的 ortholog 并不可靠,里面有太多的不确定因素会干扰结果的准确性,这也是它和基因组最大的区别所在。所以我有一段时间一度在怀疑自己这么做到底是不是对的(菜鸟就是年轻气盛啊!)。
引言
首先,研究者肯定了基因组在系统发育分析中的优越性,但也指出其成本依然昂贵,相比之下相当便宜的转录组已经成为更多研究者的选择,不过转录组依然具有以下缺点:
- 基因表达在组织间具有差异性(例如脑组织和肌肉组织所表达的基因可能有所不同)。
- 基因表达在物种间具有差异性(例如不同物种的同一基因可能具有不同的表达)。
- 高表达基因往往进化较为缓慢,而这一类基因在转录组中是大量出现的(也有其他观点认为相较于整个基因组背景而言这些编码序列进化速度更快,因为可能包括正在快速进化如经历正选择的基因)。
文章提到的这三个缺点:①、导致不知道使用哪一种组织会有更好的系统发育分析性能;②、会阻碍直系同源基因的鉴定;③、数据不完整,有很多的基因并没有被挖掘;④、进化缓慢的基因对于研究系统发育关系近的物种而言分辨率低。
考虑到这些问题,为了评估系统发育转录组的性能,研究者选择了 22 种哺乳动物和 15 种植物的转录组数据进行分析,通过比较 PT (Phylotranscriptomics) 树和 PG (Phylogenomics) 树的拓扑结构,结果表明只要直系同源基因的鉴定足够严谨,那么 PT 树和 PG 树基本一致。
方法
PG 树的构建方法:使用 OrthoMCL 从基因组序列中寻找一对一直系同源基因,比对修剪串联后使用 RAxML 推断 ML 树。
PT 树的构建方法:分成三种,分别是
- 使用 HaMStR 构建核心直系同源基因并寻找目标物种的直系同源基因,然后挑选那些含有至少 50% 物种的一对一直系同源基因。
- 基于基因树的直系同源基因推理方法。本文使用了两种方法,一种是由 Yang & Smith 提出的经验性方法(YS),一种是 PhyloPypruner。
- Orthograph 寻找(相当于是 pHMMs 的最优双向命中)。
结果
在哺乳动物中,HaMStR 和 YS 方法得到了不同的树,其中后者在拓扑结构上与 PG 树高度相似,前者就有较多的错误。基于拓扑距离得到的结论一致。
这可能是因为直系同源基因的错误识别,其中 PG 方法中错误识别的比例最小(1.9%),YS 方法的较高(5.5%),HaMStR 方法最高(28.0%)。
此外,HaMStR 鉴定得到的直系同源基因比对之后具有更高的碱基位点比例。
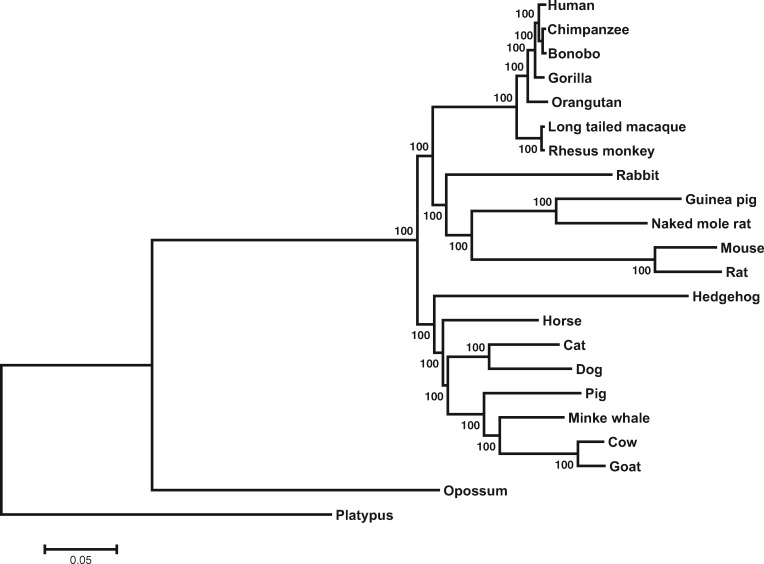
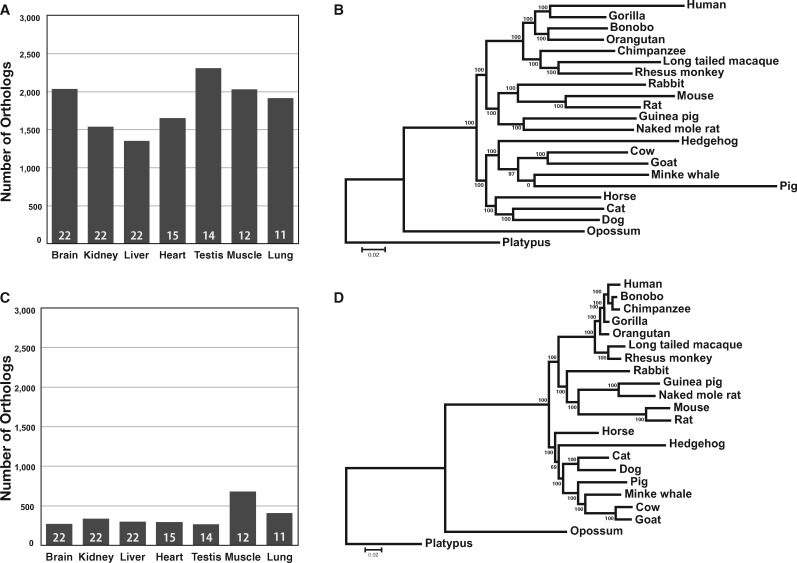
此外研究也发现使用不同的组织并不会显著降低 Phylotranscriptomics 的准确性。
为了验证这些结论的普适性,继续对植物进行了相同的操作。这次 YS 方法得到的树拓扑结构与 PG 树一致,HaMStR 树中有一个物种的位置出错。
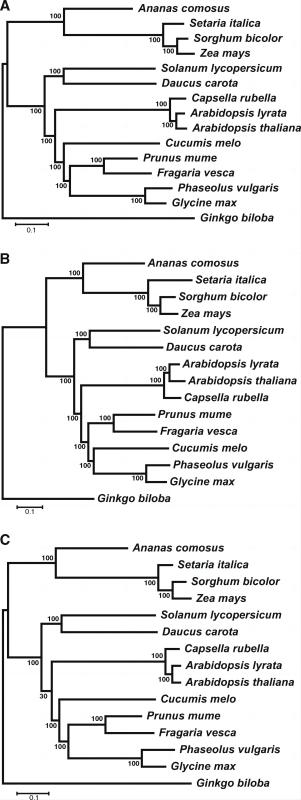
此后,为了验证结果的稳健性,使用 PhyloPypruner 和 Orthograph 进行了 PT 树构建,并且将上述操作中 OrthoMCL 进行的部分替换成 Orthofinder 又进行了一次。结果有:
- PhyloPypruner 得到了与 YS 方法几乎相同的结果(都和 PG 树很像)。Orthograph 的表现则不佳,甚至表现的还不如 HaMStR。
- 使用 Orthofinder 得到的结果和 OrthoMCL 没有太大出入。
- 不同的建树方法似乎能让某些情况下 PT 树和 PG 树的差异变小(将 RAxML 替换成 IQ-TREE 后有三个组织的 PT 树和 PG 树更像了)。
- 推断物种树的方法变换后结果依旧是相似的(串联建树 and ASTRAL-III)。
讨论
总之,本研究发现 Phylotranscriptomics 和 Phylogenomics 是一样可靠的,这有望解决以下问题:
尽管基因组数据越来越多,但是一些基于基因组的研究却也得出了相互矛盾的结果,这时更广泛的物种采样可能会有所帮助。采用 Phylotranscriptomics 时,每个物种成本的降低有助于更大规模采样的进行,这可以帮助解释一些比较有争议的树。
但是有以下那么些点需要注意:
- Phylotranscriptomics 的结果可靠性非常依赖于严格精确的直系同源基因鉴定,虽然 YS 方法鉴定的直系同源基因比 HaMStR 少一个数量级,但是显然前者的结果要可靠许多。
- 可用于系统发育推断的直系同源基因数量将随着物种数的增加而减少,这也会影响到结果的精准性。例如研究分析了 22 种哺乳动物的两个子集,它们分别包含了 14 和 7 个物种,最后得到的直系同源基因数量则依次为 235 个、705 个和 1991 个,而与 PG 树的拓扑距离则分别为 4、6 和 0。
- HaMStR 的直系同源基因识别受到所用核心直系同源基因的影响,因此使用与目标物种匹配的核心直系同源基因可能更有利于精确的系统发育推断。
后记
最后再做一些补充吧:
- HaMStR 是 RBH (reciprocal best blast hit) 方法的改进版,它的大体操作是先从核心物种中提取直系同源基因并构建隐马尔可夫模型,然后据此搜索目标物种,如果目标物种有序列能够被搜索到,那么该序列会被用来进行验证,验证的方法就是通过 RBH 看它的最佳 blast 命中是否为该直系同源基因家族中的成员,如此就能整合出包含多个物种的直系同源基因集。当然它的准确率也比单纯的 RBH 要高。
- 如果用的是
Orthofinder + PhyloPypruner的话,就算物种数很大只要跑的时间够长也是能凑出几百个直系同源基因的,不过有可能部分物种的 gap 率会比较高。 - PhyloPypruner 性能和 YS 方法差不多,但是操作上要更简单。
