比较转录组分析(六)—— GO 富集分析与可视化
前言
GO 富集分析的用处非常广泛,在这里我们将用来做差异表达基因的分析,在其他情况下也有可能是做例如正选择基因的富集分析、高突变率基因的富集分析、基因家族的富集分析等等一系列基因群的功能分析,不过着重点不同。
更新日志
2022.12.22 更改了绘图脚本,新的气泡图以 GeneRatio 作为 x 轴标签(更改版本在 github 可见,文中未修改)。
2023.06.26 解封文章,简化了部分内容。
出现的一些词
DAVID:全称 Database for Annotation, Visualization and Integrated Discovery,是一个广泛使用的富集分析平台,此外还可以在这里进行诸多 id 的转换。
富集分析过程
需要明白的概念
什么是富集分析?关于这一点在网上的诸多文章中都有解释,但我想要以一个更为通俗易懂的说法去进行解释。
打个比方,我们调查了 5000 个人的兴趣爱好,其中有 200 个人都很喜欢玩电脑。
然后我们挑出了一个我们所感兴趣的人群,比方说我们从这 5000 个人中挑出来 100 个宅。
然后我们发现在这 100 个宅中,竟然有高达 50 个人喜欢玩电脑。
于是乎,在我们的调查的所有人(即背景集)中,喜欢玩电脑的只有 200/5000 = 1/25 = 4%。
但在我们所感兴趣的人群(即前景集)中,喜欢玩电脑的就已经达到 50/100 = 1/2 = 50%。
所以我们可以认为,我们感兴趣的人群 —— 宅,他们相较于所有人群普遍而言,会更喜欢玩电脑。
需要准备的文件
无参转录组需要的文件有:
- 前景基因集(在这里是差异表达基因列表)
- 背景基因注释集(在这里是组装的注释)
如果后续需要使用 R 进行则还需要(本文的教程源自 【生信小课堂】非模式物种或无参转录组GO注释与富集 from 知乎):
- GO term 的信息(下载地址:http://geneontology.org/docs/download-ontology/#subsets 里的 go-basic.obo)
1 | wget http://current.geneontology.org/ontology/go-basic.obo |
然后使用脚本将其转化为 GO term list:
1 | #get_go_term.py |
1 | python get_go_term.py go-basic.obo |
然后就能得到 GO term list,或者你可以在我的 github 上直接下载(版本不是最新,慎重考虑),传送门点这里。
此后我们可以通过 R 或者 DAVID 进行富集分析。
通过 R 实现 —— clusterProfiler
主要是通过 R 的 ClusterProfiler 包进行,因此在进行分析前需要把这个包装好:
1 | # R version 3.5以上版本: |
具体的安装教程可以 Bing 搜索,安装好后就可以在 R 上进行后续的分析,也可以在装了 R 的服务器上进行。
用自己电脑的 R 画图是完全可以的,但如果涉及到的要分析的物种比较多时就会显得比较麻烦,所以在这里我制作了一个 python pipeline 在服务器上运行直接得到想要的结果:
1 | # -*- coding: utf-8 -*- |
脚本的下载可以去 github,如果无法在服务器上使用 clusterProfiler 也可以把中间那一段 R 代码抄下来在自己的电脑上运行,只需要换掉一些特定的字符即可。
运行例 ①:
1 | python enrichment_plot.py -gef all_up.txt \ |
使用所必须调用的参数:
-gef 分析使用的前景基因集,例如上调或下调的差异表达基因。
-geb 分析使用的背景基因注释集,即之前文章中所得到的一基因对一 GO term 文件。
-go 在需要准备的文件中有提及到的 go_term.list。
-o 输出文件夹。
运行例 ②:
1 | python enrichment_plot.py -c enrichment_analysis.csv \ |
当使用已有的 csv 进行分析时只需调用该 csv 文件即可(列名必须与运行例 ① 中输出的保持一致)。
在该情况下只会输出对应的图及原始 R 代码,不会再次输出 csv 文件。
可选参数:
-t 图的标题,默认不设标题;-l -w 图的高和宽,默认都为 8;-y 当为 ID 时,作图的 y 轴标签会替换为 GO 号,默认为 Description 。
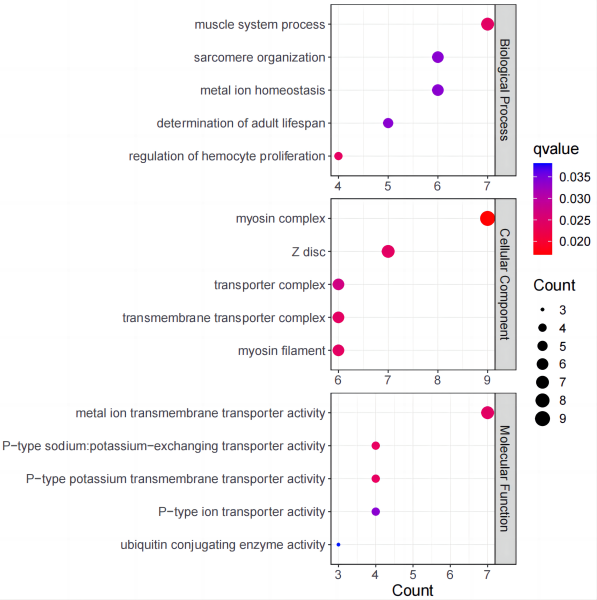
以运行例 ① 为标准,运行结束后,会在 outputdir 中出现一个以运行时间作为名字的文件夹,里面包括了以下几个文件:

enrichment_analysis.csv 为富集分析的结果,其中已被废弃的 NA 项未被去除(运行例 ② 不会输出该文件)。
enrichment_plot.pdf 为富集分析的气泡图,其中去除了 NA 项进行分析,取每个 Ontology 前五显著的 GO term 进行展示(不足五个则取全部)。
enrichment_plot_top10.pdf 为富集分析的气泡图,其中去除了 NA 项进行分析,取最显著的十个 GO term 进行了展示(不分 Ontology)。
enrichment_plot.R 为该次分析的代码。

另:曾在 比较转录组分析(四)—— 组装的 GO 注释 中提到过,富集分析里可以考虑把一些相关的 term 给删掉一部分,仅保留具有代表性的那个进行可视化,这里可以基于运行例 ① 输出的表格进行修改,删除一些相关 GO 项后再通过运行例 ② 的方法直接可视化。
通过 DAVID 实现
DAVID 的分析会严格很多,可能会导致我们得不到什么想要的结果,但是这种严格又未必不是件好事。
首先 DAVID 的 Document 可以看这里:DAVID Bioinformatics Resources (ncifcrf.gov)。
为什么说 DAVID 严格?因为它在进行分析的时候首先把重复项删除了,其次还把所有原始 ID 中的冗余给去除了,于是乎很多原来在 clusterProfiler 的分析中具有富集现象的 term 在 DAVID 就失去了显著性。
这里我就以上面 clusterProfiler 中使用的数据为例,分享如何利用 DAVID 进行 GO 富集分析。
首先,我们要提取出原来我们 blast against swissprot 得到的注释信息(blastx.outfmt6):
1 | cut -f 2 blastx.outfmt6 > back.txt |
得到背景基因的注释集后,我们再通过上调基因和下调基因的列表,得到背景基因注释集:
1 | cat all_up.txt | while read line; |
有了背景基因和前景基因注释信息后,就可以把它输入进 DAVID 进行分析。

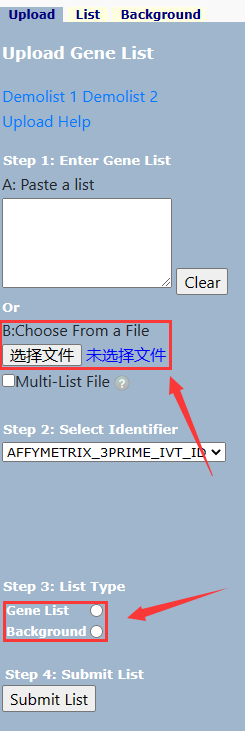
Gene List 输入上调或下调的差异表达基因集,Background 输入背景基因注释集,它们是这样的(一行一注释):

在 Select Identifier 上选择 Uniprot ID。
它会提示检测到基因集中有很多不同的物种,略过即可。
弄完以后点击右侧 Step2 下面的内容进入下一步分析,如果只需要做 GO 富集分析,则可以先按 clear all,然后勾选这三项:
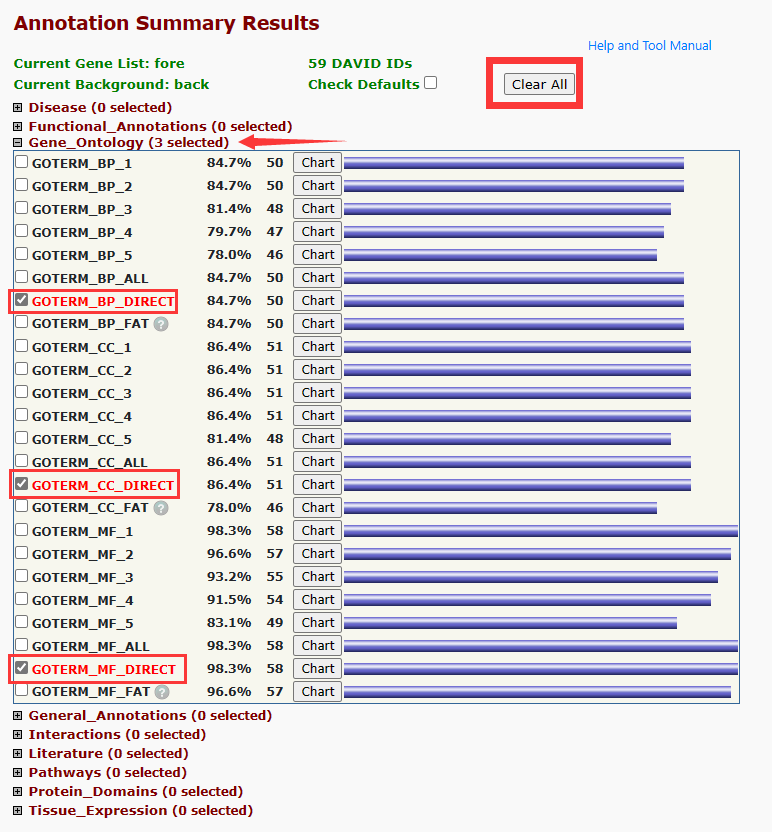
然后移到最下面有三个选项,其中 Functional Annotation Chart 就是 GO 富集结果了。
DAVID 采取的检验方法为费希尔精确检验,基于对应的 DAVID ID 进行计算 p 值和对应矫正值。以该文中涉及的数据为例,显著富集的 term 如果要以 Benjamini 矫正值为准,那么显著富集的 term 仅有三个,远远低于在 clusterProfiler 中识别到的 44 个。
后续的可视化步骤同样用 R ggplot2 实现,可以参考上文的代码进行。
后记
下一篇预告:比较转录组分析(七) —— 系统发育分析。
可能会出的:基于 Cytoscape 的富集网络图实现。
如果有时间的话...... —— Juse
关于 BH 矫正
我不打算用过于科学的说法来解释做这个的动机,所以我找了一个很有意思的图片来展示:

我再来说一段当初在生物统计学课上听老师讲的故事(内容可能有差异但大致相似):
有一只被称作神奇章鱼的章鱼,在进行拳击比赛时让它进行猜测谁会赢,结果在二十次比赛里有十九次它的触手都伸到了胜利方的牌子上,也就是说二十次它猜对了十九次,大家觉得这只章鱼是不是真的是一只具有魔力的、神奇的章鱼?
大家都会觉得比较神奇吧,19/20,95% 的正确率。
但是我再跟大家说一个事,事实上这个世界上有数千数万只章鱼正在进行这种猜测,但是只有几只章鱼脱颖而出,那么这时候大家还会觉得这是一只神奇的章鱼吗?
这个故事精准地切中了多重比较中存在的问题,也就是子集和整体之间的问题,一个事件的发生概率再怎么样低,如果它重复的次数多了,那么它总会在一次试验中发生并在那次试验中展现出显著性,但实际上放在整体上看如果相信了这个显著性就会给我们带来错误的结论,这也就是我们要进行 p 值矫正的原因。
BH 矫正的办法就是:adjust.p = p * n / k
其中,p 是指原来的 p 值,n 是指这次矫正中有 n 个 p 值,k 是指将 p 值从小到大排列以后该 p 的顺序数。
当排在前面的 p 值在矫正后比在后面的 p 值大时,排在前面的 p 值又会重新矫正为和后面 p 值相同的值,这也就是在结果中经常能看到好几个矫正后的 p 值都相同的原因。
clusterProfiler 在 linux R 上的安装
这里简单阐述下我自己是如何解决这个问题的:
首先,每个 R 包都可以通过 conda 安装,可以在 Bing 上搜索关键词 xxx conda。
例如可以先搜索: clusterProfiler conda

点进去,选择上方的 Files,在里面看到不同版本的以及适应不同平台的 R 包文件。
如果在 linux 系统上安装,则选择开头为 noarch 或者 linux 的文件下载。
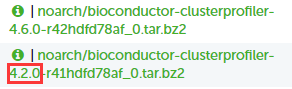
此外,如上图红框所标注为该版本 R 包构建时所基于的 R 版本,因此还需要根据自己的 R 版本选择合适的包。
例如如果自己的 R 版本是 4.1 的,那么这里就可以选一个 4.0 的下载安装。
此后在 linux 中输入下列命令用 conda 安装 R 包:
1 | conda install xxx.tar.bz2 |
这样做仅是把这个包给安装上了,但是众多的依赖项依然没有完备,因此在安装好后进入 R 并尝试加载时,系统会提示缺少什么包。
此后把缺少的包一个一个用上述方式搜索然后再用 conda 安装,直到依赖项全部弄好为止。
当然,也可以尝试直接用 R 的 BiocManager::install 安装,但并不总能成功。
此外,如果安装某个依赖项以后在加载 clusterProfiler 时依然显示缺失该依赖项,换一个依赖项的版本重新安装可能能够解决这个问题。
全部安装后以后,再进行一次就不会报错了:
之前我束手无策时选择了这条道路,虽然很笨,但是管用。