比较转录组分析(五)—— 转录本定量与差异表达分析
前言
之前看知乎看到过一篇关于写生物论文的文章,里面有一段很有意思的话,具体的内容我已经忘了,但意思大致是指写生物论文最好 “水” 的就是差异表达分析,我看到的第一反应是乐,第二反应是这可能确实是客观存在的。
不过会的人多用的人多并不代表这个分析烂大街没有含金量,相反,这么多人用应当是证实了这一分析的价值所在。
更新日志
2022.12.12 在 DEG 挑选部分中补充了些关于使用 voom 的内容。
2022.12.21 更改了关于 log2FoldChange 阈值的说法。
2022.12.22 补充了题外话中关于软件差异的新发现。
2023.04.20 补充了题外话中关于软件差异的新发现。
2023.07.03 精简了内容。
出现的一些词
log2FoldChange:差异倍数,常以 1 作为阈值,有时也可为 1.2 或 1.5(一些单细胞研究中常见这些值)。
例:若对照组表达量为 1,处理组表达量为 4,则 log2FC = log2(4/1) = 2。正数时为上调,负数时为下调。
生物学重复:经过相同方式处理的不同样品,提高生物学重复可以显著提高检测结果的准确性,一般而言一个较为可靠的分析需要每个处理至少三个重复。
例:3 个小鼠放在低温下处理,3 个小鼠放在常温下处理,然后比较不同温度下的小鼠转录组差异。
关于生物学重复的重要性可以看看这篇综述:
Todd, Erica V et al. “The power and promise of RNA-seq in ecology and evolution.” Molecular ecology vol. 25,6 (2016): 1224-41. doi:10.1111/mec.13526
原始表达矩阵:进行差异表达分析时需要用到的各基因表达矩阵,里面记录了每个测序数据对应的基因表达量,被称为原始是因为还没进行标准化(不同测序数据的测序深度可能不同,会导致整体表达量的差异),标准化后可减弱测序深度差异带来的影响。
探索性研究(Exploratory research)和描述性研究(Descriptive Research)的差别:
- Exploratory research is one which aims at providing insights into and an understanding of the problem faced by the researcher. Descriptive research, on the other hand, aims at describing something, mainly functions and characteristics.
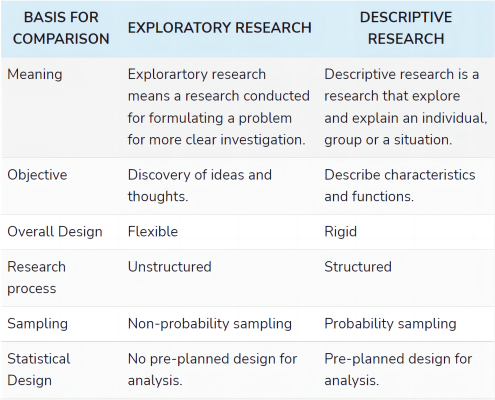
- Conclusion: Therefore exploratory research results in insights or hypothesis, regardless of the method adopted, the most important thing is that it should remain flexible so that all the facets of the problem can be studied, as and when they arise. Conversely, descriptive research is a comparative design which is prepared according to the study and resources available. Such study minimises bias and maximises reliability.
Reference: Difference Between Exploratory and Descriptive Research (with Comparison Chart) - Key Differences
批次效应:不同时间、不同操作者、不同试剂、不同仪器导致的实验误差。
如果批次效应很严重,就可能会和真实的生物学差异相混淆,让结果难以捉摸。
差异表达分析过程
可以看到很多用于进行差异表达分析的比对软件,但这里首先要着重说明 Hisat2 这个软件是用于 有参 情况的。而对于无参转录组而言主流的比对软件是 Bowtie2 ,不过这两个软件也同样是由同一队人开发。
具体的实现将依靠 Trinity 成熟的内置脚本进行,教程可见 https://github.com/trinityrnaseq/trinityrnaseq/wiki/Trinity-Transcript-Quantification ,这里主要就是把它翻成中文。
关于 Trinity 的安装,你可以使用 conda 进行(或者 mamba):
1 | conda install bioconda::trinity |
下文中使用到的 Trinity 内置脚本,如果不清楚其位置可以在安装 Trinity 后使用 locate 命令寻找,例如:
1 | locate align_and_estimate_abundance.pl |
请注意 locate 需要在新文件产生一段时间后才能定位到,且部分内置脚本在早期的 Trinity 中并不存在(该文章使用的版本为 2.13.2)。
估计转录本丰度
使用的脚本是 Trinity 的 align_and_estimate_abundance.pl,不知道位置可以使用 locate 命令查找(没找到可能是版本不够)。
1 | nohup align_and_estimate_abundance.pl --transcripts cdhit.fasta \ |
此处 nohup 用于避免终端意外退出导致运行中断,如果服务器或主机没有该程序也可去除,在这里:
--transcripts为转录组组装的位置。--seqType用 fq 就行。--samples_file在组装的时候我们用到过,如果不用该参数则替换成相应的--left和--right。
tip:如果在组装时使用过 --trimmomatic ,那么这里的双端数据应该替换成质控后的位置,此外,也有可能参考转录组的组装数据并不是用来做差异表达分析的,因此可以直接在 --transcripts 指定以后,sample.txt 中放进行差异表达分析的数据就行。
在这里再放一下 sample.txt 所应该有的格式:
1 | cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq |
可把第一列看成不同的处理,第二列看成每个处理的生物学重复,第三第四列是对应的双端文件(也可为单端)。
--est_method是丰度估计方式,分三个软件,有RSEMkallisto和salmon,这里用RSEM。--aln_method是--est_method选择RSEM时需要配置的,可以选择bowtie和bowtie2,此处使用后者。--thread_count线程数,越大越快,但大到某个值以后再加效果有限,所以不必用太多。--debug运行时开启该参数,在运行结束后不会删除中间文件,如果所剩空间不多可以不用加这个参数。--prep_reference自动为转录组建立索引。--SS_lib_type当测序数据为链特异性时需要添加,否则会导致定量不准确。>bowtie.out 2>&1最后这一行是为了将运行过程中的屏幕输出储存到 bowtie.out 这个文件中,方便后续查看比对率等信息。
运行结束以后,会跑出来所有的比对结果,以上述 sample.txt 为例,跑出来的各个文件应该长下面这样:
1 | ls |
跑出来的都是文件夹,里面的 RSEM.isoforms.results 就是各个重复的定量结果。
然后,可以用一串命令将这些定量结果统计起来:
1 | ls */RSEM.isoforms.results > countfile.txt |
所有的定量结果位置都已统计在 countfile.txt 中,此后运行下列命令:
1 | abundance_estimates_to_matrix.pl --est_method RSEM \ |
此处因为我们选择在 isoform 水平上分析,因此 --gene_trans_map 选用 none。
使用 --name_sample_by_basedir 以让表达矩阵中的样本名与文件夹一致。
运行结束后得到的 RSEM.isoform.counts.matrix 就是我们在进行差异表达分析时需要用到的原始表达矩阵了。
接着我们再运行:
1 | cut -f 1,2 sample.txt > sample5.txt |
这里的 sample5.txt 可用于后续的样本质量检查和差异表达分析。
检查样本质量
如果觉得需要进行:https://github.com/trinityrnaseq/trinityrnaseq/wiki/QC-Samples-and-Biological-Replicates
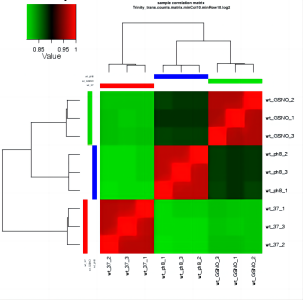
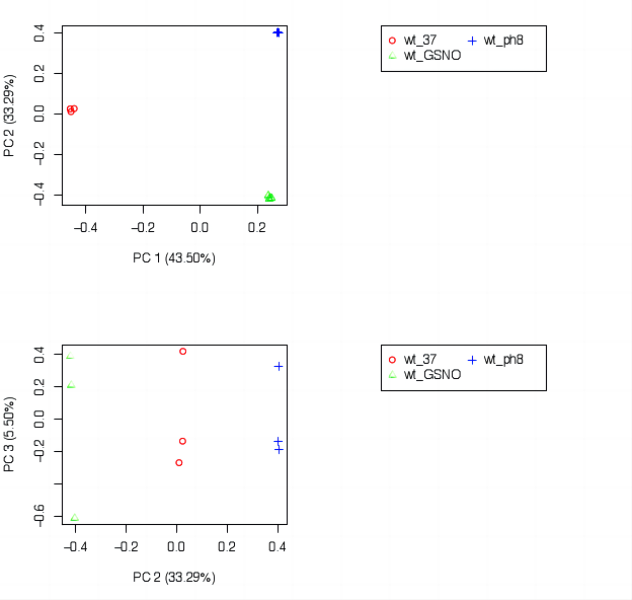
我个人觉得所有图里最有价值的是 sample_cor_matrix 和 principal_components 的图(相关性热图 & 主成分分析)。
运行基本都是照官网教的做,而且这个脚本的可设置参数非常多,里面的某些默认参数似乎需要根据自己的数据情况进行微调才更严谨。
这一步最好还是跑一下,正常的情况应该是:
①、在 sample_cor_matrix 中,不同处理的各个生物学重复应该要分别聚在一块。
②、在 principal_components 中,一个处理的各个生物学重复的点应该在邻近的区域内,不同处理的点应该要分开。
例子:
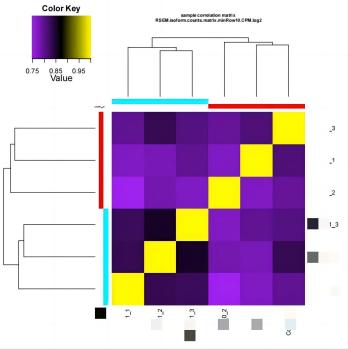
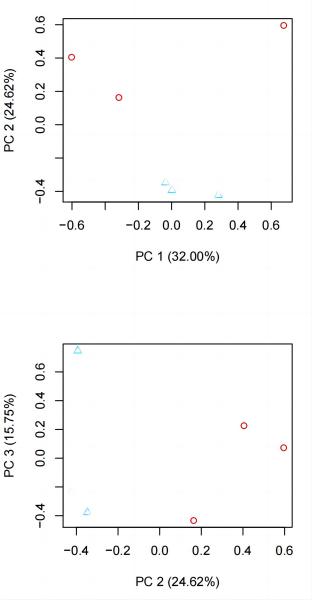
如果不同处理混到一起了有可能是下面的几个情况之一:
①、样本的变异系数很大,也就是说这个生物即使是在同一处理下,不同个体之间也可能表现出较大的差异,所以彼此之间很离散。
②、实验方法上的问题,可能样本处理不当等;又或者是样本编号错误。
如果同一处理下有很明显分开的两个区块则可能是:
①、批次效应,可见 批次效应 from 简书。
②、巧合。
差异表达分析
可选择的软件:edgeR、DESeq2、limma-voom。
前两者的使用应该是最广泛的,具体使用哪一个可以根据具体情况确定,例如:
①、如果差异表达基因鉴定数量较少,可以选择得到更多差异表达基因的软件,亦或者是取多个软件的并集。
②、如果差异表达基因鉴定数量很多,可以取多个软件的交集。
关于取并集和交集,两个方法都有人用,探索性(exploratory)的研究用前者也行,但如果是描述性(descriptive)的研究用后者会更合适,因为总的来说肯定是后者更为精准可靠(假阳性率更低)。
首先,要先检查一下自己的 linux 中是否有所需要的包,不确定可以直接安装试试:
1 | R #进入 R |
安装好以后输出 q() 退出 R,并使用 Trinity 的 pipeline 进行分析,首先你需要这样一份文件:
1 | cond_A cond_B |
中间是制表符分列,命名成 contrast.txt。可以是多行也可以是一行,里面不同的 cond 代表不同的处理,每一行代表一次比对,排在后一列的是对照组。
以上述文本为例,后续的差异表达分析将以 cond_B 作为对照组,以 cond_A 和 cond_C 作为处理组分别进行两次比对并输出结果。
记住处理组和对照组千万不要放反了,不然后续的分析会全部按照真实情况的相反面进行,我依然记得在之前生信课上有一个小组汇报的很好,但是由于这一步做错了她们把上调基因当下调分析,下调基因当上调分析,虽然听她们讲的很好但是事实上完全是反着来讲的。
将 contrast.txt 弄好后运行下列命令:
1 | DifferentialExpression/run_DE_analysis.pl \ # 老样子,不知道位置可以用 locate 确定。 |
--matrix一定要用原始表达矩阵!每个软件都有它自己的标准化步骤嵌在分析流程中,所以不需要担心。--method有DESeq2edgeR和limma-voom。--min_reps_min_cpm筛选基因,例如此处的意思是筛选至少在三个样本中出现的且 count-per-millon 不低于 1 的基因。
运行结束后会得到一个表格,此后可以用这个表格进行火山图绘制或挑选差异表达基因。
DEG 挑选
在差异表达分析结果中以 |log2FC| > 1, q-value (or FDR) < 0.05 作为阈值进行筛选。
挑选方法:自己手动在 excel 里面筛选或者用 python:
1 | import sys |
这个我针对上述脚本输出的文件所写的一个提取差异表达基因的脚本,如果想要更改筛选标准可以在里面做相应的调整,或者添加可指定的参数项。
使用例:
1 | python de.py xxx.DE_results DEG_up.txt DEG_down.txt |
输出的 DEG_up.txt 和 DEG_down.txt 里分别是上调和下调的差异表达基因列表(可根据自身需求调整输出的文件名称),后面几步可选择进行,用处主要是合并所有软件得到的差异表达基因以及统计有多少上调和下调的基因。
获得差异表达基因以后,就可以准备进行后续的富集分析了。
后记
下一篇预告:比较转录组分析(六)—— GO 富集分析与可视化。
火山图教程预告(随缘):基于 GraphPad Prism 的火山图绘制。
使用 R 绘制火山图的话可以看看这一篇教程:R 数据可视化 02 | 火山图 from 知乎。
①、关于三个转录本丰度估计软件的差异
RSEM 是需要比对结果进行定量的,可以调用的软件有 STAR Bowtie2 Bowtie 等,文里使用了 Bowtie2。其他两个是不需要进行比对就可以直接进行定量的工具。具体哪个最为准确的问题,感兴趣可以看一下这篇论文:Interoperable RNA-Seq analysis in the cloud。这篇文章对 salmon 和 kallisto 评价是比较高的(不过和 qPCR 的结果最不相似,事实上和其他的也就差了一丢丢,我个人不排除误差问题),RSEM 的结果和它们相似但是由于运行效能的问题被 diss 了。
再找,然后在 kallisto 的论文中找到了这张图:
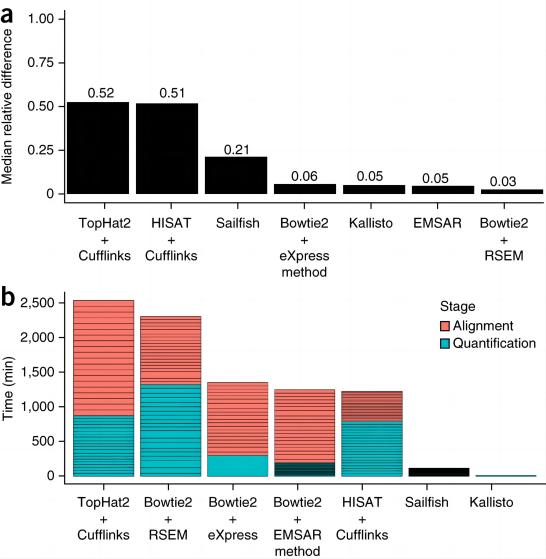
简单来说就是把每个转录本定量结果的中位数差异作为精确性的指标,然后可以看到 Bowtie2 + RSEM 的组合是最为精准的,kallisto 虽然次了一点点但也不遑多让。而当考虑到运行时间时,Bowtie2 + RSEM 的组合很不幸地被 kallisto 碾压了,还不是一点点的程度。可以看到 kallisto 的定量是非常迅速且也保证了精确度的。所以运行内存有限并且追求精确度的情况下,kallisto 毫无疑问是更好的选择。
此外在一篇讨论 salmon 精度的文章中,Bowtie2 + RSEM 在某些情况下的精准度比其高(当数据和参考转录组一致的情况下),当参考转录组和 reads 并不完全匹配时,则 salmon 更为精确些。
文章:Alignment and mapping methodology influence transcript abundance estimation
关于不同软件具体的差异在哪里可以精读一下文章,这里我就只搬个结论作为参考。
软件的效率(速度)是一个非常重要的参考依据,如果想用有限的内存得到最好的效果,那么 kallisto 和 salmon 的优先级要高很多。
②、不同差异表达分析软件的差异
找文章,直接找文章:
from Genome Biology
这里就不放图了,简单说一下几个结论:
①、在大样本量时,DESeq2 和 edgeR 具有惊人的假阳性率,两者中 edgeR 更为明显。
造成这些现象的原因是:However, while the three parametric methods were initially designed to address the small-sample-size issue, these population-level studies had much larger sample sizes (at least dozens) and thus no longer needed restrictive parametric assumptions. Moreover, violation of parametric assumptions would lead to ill-behaved p-values and likely failed FDR control, an issue independent of the sample size.
简单来说,就是这三种方法都有设置相应的假设以应对低样本量的情况(这个假设允许混合有相近表达情况的基因数据以估计方差),然而在大样本量的研究中这一假设不再被需要,如果继续坚持这一假设会导致错误的估计。
②、相比之下大多数情况中 limma-voom 表现更为良好(不管小样本量大样本量)。
③、推荐在大样本量时使用 wilcoxon 秩和检验,这种非参数的方法在面对异常值时会显得更加可靠。但与此同时,由于其是一种非基于回归的方法,因此无法针对混杂的因素进行调整,因此使用该方法进行 DEG 鉴定前需先标准化或者使用概率指数模型调整协变量。(文中的方法是先用 edgeR 进行过滤和标准化然后再使用 wilcoxon 秩和检验 + Benjamini&Hochberg 矫正)
tip: 有趣的是我发现网上有关于第一篇文章的介绍,写的非常好,传送门:别再用 DEseq2 和 edgeR 进行大样本差异表达基因分析了。
补充(2023.04.20):
事实上我在大概一月份左右就发现了新东西,但是一直没有更新,今天心血来潮突然就想起来,于是乎就进行了补充。
新的发现就是:
样本多(重复多)时,edgeR 多于 DESeq2。
样本少(重复少)时,edgeR 少于 DESeq2。
这可能与它们的算法差异有关,这一点可以看 DESeq2 论文原文片段:
Besides the need to account for the specifics of count data, such as non-normality and a dependence of the variance on the mean, a core challenge is the small number of samples in typical HTS experiments – often as few as two or three replicates per condition. Inferential methods that treat each gene separately suffer here from lack of power, due to the high uncertainty of within-group variance estimates. In high-throughput assays, this limitation can be overcome by pooling information across genes, specifically, by exploiting assumptions about the similarity of the variances of different genes measured in the same experiment.
Many methods for differential expression analysis of RNA-seq data perform such information sharing across genes for variance (or, equivalently, dispersion) estimation. edgeR moderates the dispersion estimate for each gene toward a common estimate across all genes, or toward a local estimate from genes with similar expression strength, using a weighted conditional likelihood. Our DESeq method detects and corrects dispersion estimates that are too low through modeling of the dependence of the dispersion on the average expression strength over all samples.
③、关于批次效应
一个会毁坏整个实验的事情。
事实往往比言语更有力:
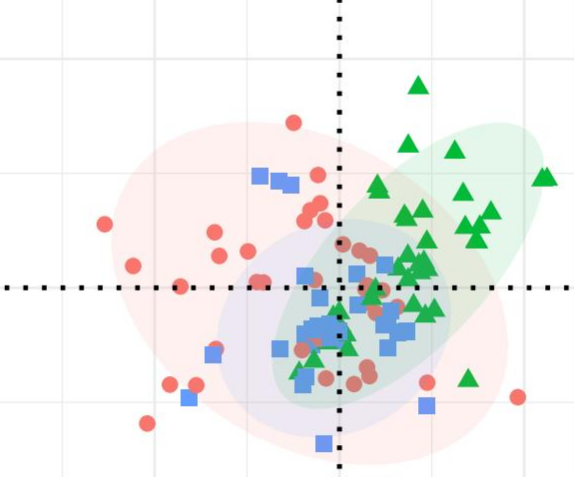
这是一张 NMDS 图,乍一看好像有挺多值得琢磨的东西,比如这三组里面红色组和绿色组的势头是不一样的。
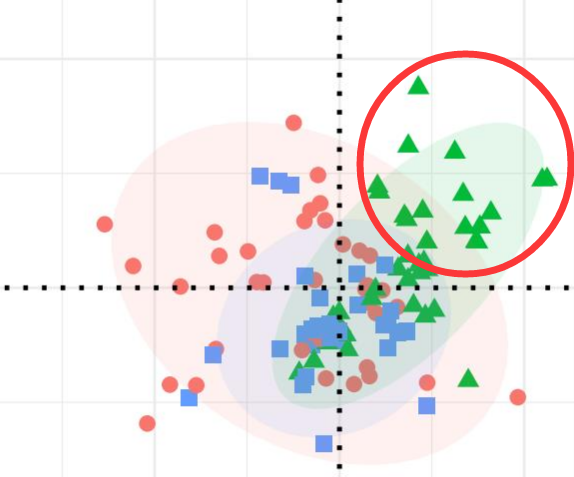
然而事实上,红圈所框住的地方所对应的样本与其他样本的采集时间是不一致的(相隔了两个季节),导致虽然这些样本的分组和同组其他样本是一样的,但它们的 NMDS 结果却截然不同并且出现分层。
这就是批次效应,也是很多研究者们想要尽力避免的问题。所以在设计实验时就应当注意,需要把除了自己关注的因素以外的所有其他干扰因素给排除或者尽力降到最低,避免出现部分结论无法解释的现象以及避免产生批次效应。