比较转录组分析(四)—— 组装的 GO 及 KEGG 注释
前言
之前我们已经讨论了评估组装质量和组装去冗余的方法,在这里我们再来进一步谈一谈组装的注释。
这一环节应该算得上是转录组的第一个重头分析部分,因为它可以让执行者对自己的组装有一个更清晰的认知。
更新日志
2022.12.29 补充了如何利用 GO 注释信息绘制二级分类图的部分。
2023.01.11 补充了如何利用 Kofamscan 进行 KEGG 注释。
2023.07.03 精简了部分内容。
出现的一些词
blast:中文名叫做爆炸,其实是一种比对工具。
diamond:中文名叫做钻石,其实也是一种序列比对工具,速度是 blast 的 100-10000 倍,且资源需求低。
swissprot:一个注释非常良好的蛋白数据库,是 UniProt 数据库的组成之一。
KEGG:全名京都基因与基因组百科全书,由日本京都大学生物信息学中心搭建的一个数据库。
什么是注释
注释分很多种,但是它们在转录组分析中的最终目的都指向一个共同点 —— 解释组装(或者说基因)。
比如说 GO 注释、KEGG 注释、Pfam 注释、nr 注释的注释内容是不一样的,但是最终目的都是想阐明组装中不同的 contig 代表的是哪一种基因或者说执行了什么生物学功能。
不过注释库都是人为编写的,并且对于每个基因的注释也都是人为划定的,关于这一点有很多可深入了解的地方,关于 GO 注释可以看一看这两篇文章:GO注释和富集分析概念介绍 from 简书 与 Gene Ontology(GO)简介与使用介绍 from 知乎。
GO 注释
几点可能需要注意的事情:
①、GO 注释被分为三大类,分别是生物过程、细胞组分和分子功能。
②、GO terms 之间可能是有关系的,比如一个 term 是另外一个 term 的父级项,整个 GO 的结构是一个有向无环图。
关于第 ② 点,我随便举个例子,比如说
吃饭和吃下午饭这两个 term 里,明显可以看出吃饭是吃下午饭的父项,因此在某个基因上如果出现了吃下午饭这个 term,那么它就同时拥有吃饭这个 term。
③、基于上述原因,一个基因中可能会出现非常多相关的 GO term 注释,而在后续的富集分析中也会表现出来(比如说前几的都是一些相关联的 GO term),因此在后续的富集分析里可以考虑把一些相关的 term 给删掉一部分,仅保留具有代表性的那个。
④、GO term 的注释事实上是相对宽松的,只要这个基因跟某个功能沾上了边就会有人给它这个功能对应的注释。
⑤、GO term 是一个一直在保持更新的注释库,因此老旧的注释库数据可能存在某个 term 被废弃掉的情况,因此在某个项目的分析过程中最好保持数据库版本的一致,并在不需要继续使用的时候抽空更新数据库(记录好使用过的版本、做好版本控制,保证结果的可再现性)。
就 GO annotation 而言,可以选择的软件有很多,比如 Interproscan、eggnog-mapper、Trinotate、Blast2GO 等等,包括一些在线网站比如像 Pannzer2 等也能够完成这一任务。
值得注意的应该是 Trinotate 的开发者也开发过 Trinity ,两个软件之间的衔接非常好,甚至有一些 Trinotate 的功能就是专门为 Trinity 的组装提供的,所以后续我会就 Trinotate 的注释方法进行分享介绍。

Trinotate 安装 & 执行
更新于 2024/10/17:
经读客朋友反馈,现发现下述内容已不适用于最新版本的 Trinotate,其改动大致有如下:
①、目前的 diamond blast 方法已经被 Trinotate 采用为标准方法(旧版本中仅有 blast+)。
②、目前的 Trinotate 实现了自动化的数据库构建,无需自己进行下载(旧版本中需自行构建)。
③、总体流程有所出入,部分旧版本流程源码被废除,部分旧版本流程源码有保留但是未被其放在 wiki 中展示。
因此,建议各位朋友直接前往 Trinotate Wiki,跟随最新版本的指引进行相关安装和注释:
https://github.com/Trinotate/Trinotate/wiki/Software-installation-and-data-required
本文章的内容将保持不变,部分流程依然适用(作为参考),也在此提醒各位做好自己研究中的版本控制工作。祝各位研究顺利!
安装可以参照 Trinotate 官网进行(传送门),不过在这里提出几点可能可以帮助减少软件配置时间的建议:
①、如果仅使用 Trinotate 进行 GO 注释而不进行别的注释,那么仅安装 Software Required 中的 Trinotate、TransDecoder、SQLite 和 NCBI BLAST+ 就可以满足运行需求。
②、NCBI 的 blast 速度事实上挺慢的,如果转录组较大而且希望运行时间短一些,可以考虑使用 diamond blast 进行后续的步骤。
Trinotate 的注释原理简单来说,就是首先进行了 SwissProt 的注释,然后根据注释信息输入到 SQLite 数据库中并调用出注释蛋白对应的 GO term 注释。因此首先我们会对组装进行蛋白预测和 blast 比对:
1 | TransDecoder.LongOrfs -t cdhit.fasta |
Transdecoder 会预测组装中可能存在的开放阅读框并得到对应的 cds 序列和 pep 序列。
注意,这里应该使用去冗余后的组装作为注释的背景集。此外,TransDecoder.Predict 可使用 --single_best_only 参数使其仅输出每个 contig 预测的最佳 pep 序列和对应 cds(原理是先看同源性再看长度,但貌似大多数时候都是取最长)。
在这之后,使用 diamond blast(也可以使用 NCBI blast)against swissprot database:
1 | diamond blastx --query cdhit.fasta --db uniprot --threads 5 --max-target-seqs 1 --outfmt 6 --evalue 1e-5 > blastx.outfmt6 |
blastx 是用核苷酸序列去比对蛋白数据库,blastp 是用蛋白序列去比对蛋白数据库,这里以 evalue 为 1e-5 为阈值,按照官网的 1e-3 也没有问题不过在文章中写明就好。
得到两个 blast 文件后,使用 Trinotate 将其加载到 SQLite 数据库中并输出注释报告:
1 | Trinotate /path/to/Trinotate/Trinotate.sqlite init --transcript_fasta cdhit.fasta --transdecoder_pep transdecoder.pep --gene_trans_map cdhit.map |
tip:此处的 /path/to/Trinotate/Trinotate.sqlite 需要用户自行进行构建和指定,详细可见 Trinotate Sequence Databases Required。
注释报告中详细展示了每个 contig 的注释信息,此后可使用 Trinotate 的脚本输出为 GO 注释文件:
1 | Trinotate/util/extract_GO_assignments_from_Trinotate_xls.pl \ |
其中 -T 指的是在转录组水平上进行,可替换成 -G 在基因水平上进行,但是我们已经进行过去冗余了所以使用 -T 即可。
--include_ancestral_terms 指的是会沿着有向无环图爬取每个 GO term 的父项 term 并输出。
得到的文件是长这个样子的:
1 | xxx1 GO:0003674,GO:0005488,GO:0045216,GO:0045296 |
此后,为了后续可以使用 R clusterprofiler 进行 GO enrichment analysis,我们需要把它修正为一对一的模式,脚本如下:
1 | import argparse |
脚本我放在 github 里了(传送门),或者你可以将其复制到命名为 onego.py 的脚本中,使用方法如下:
1 | python onego.py -r go_annotation.txt -o onego.txt |
得到的 onego.txt 长这个样子:
1 | xxx1 GO:0003674 |
到这里就算获得了组装的背景基因注释集,接下来就是通过差异表达分析获得 DEGs(Differential Expressed Genes)得到前景基因,并通过富集分析判断差异表达基因的功能。
GO 二级分类柱状图绘制
一些可用的在线网站:
网址:https://www.omicshare.com/tools/Home/Soft/osgo
上述两个网站的绘图都可以进行相应的参数调整,感兴趣的友们可以自行尝试(格式可能需要进行一定的调整)。也可以自己写脚本然后统计二级分类数量并绘制柱状图。
KEGG 注释
方法
最简单的方法是通过官网进行,地址:https://www.kegg.jp/blastkoala/ ,官网中的方法分成了三种,分别是 BlastKOALA、GhostKOALA 以及 KofamKOALA。使用无参转录组进行注释的话需要注意:
- 需要使用 pep 序列(可使用 Transdecoder 预测出来的)。
- 不要使用 GhostKOALA(用于预测宏基因组),其他两个都可以。
不过官网注释有个缺陷就是它有序列数限制,因此除非我们把自己的 pep 序列分成很多份然后一份份交给它注释,否则这个办法是行不通或者说可行度很低的。
所以说更好的方式是采取本地化的注释方法 —— Kofamscan 进行。
Kofamscan 安装 & 执行
目前网上关于 Kofamscan 的教程还是非常多的,所以这里可以自行 Bing 搜索安装并尝试执行。
好教程:KEGG功能注释及本地化--基于KofamKOALA from 知乎
用于理解 KEGG 注释的好教程:一文快速读懂 KEGG 数据库与通路图 from 知乎
输入命令执行 Kofamscan :
1 | exec_annotation -o kegganno.detail.csv --cpu xx -E 1e-3 cdhit.pep |
其中把 cdhit.pep 换成自己的 pep 序列就行了。
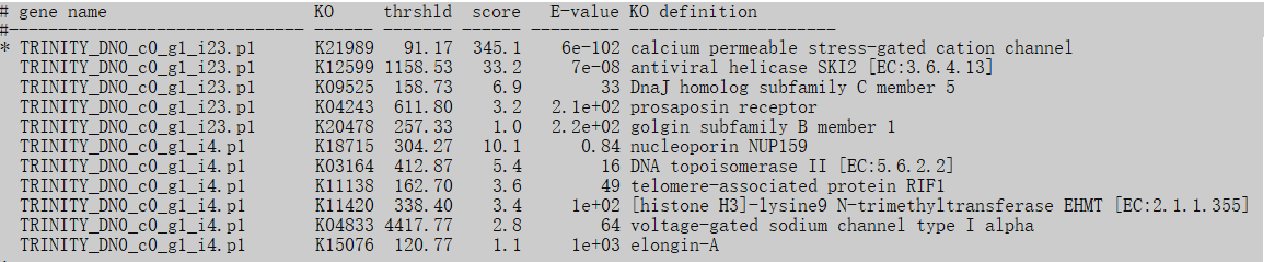
使用上述第一条命令的输出(未使用 -f mapper),会输出一份详细的注释报告,包括基因得到的 KO 号以及分数、e 值等信息,当基因名前有 * 时代表该基因的分数超过阈值,说明这个注释是可信的。
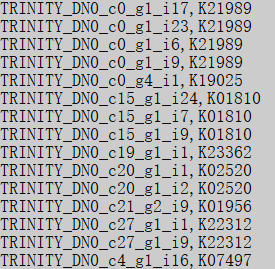
使用上述第二条命令的输出则是会把分数超过阈值且 e 值小于 1e-3 的那些注释输出成一份一基因对一 KO 的格式,这种格式方便我们处理用于后续的富集分析。
事实上 Kofamscan 的注释仅仅只是 KO 号,一般而言我们在论文里看到 KEGG 注释和 KEGG 富集分析事实上指的都是 KEGG 通路(pathway)注释和通路的富集分析,而 KO 号和 kegg 通路之间是有很大差别的。
在这里我就直接搬上面知乎教程里的讲解展示:

KO 的全称即为 KEGG ORTHOLOGY,orthology 的意思也即同源。
而在 KEGG 数据库中,代表着通路的 id 在总库里是由小写的 ko 开头的,细分到具体的物种还会有诸如 hsa 开头(代表人类)等样式的通路 id。而一个基因可能参与到多个生理过程中,因此一个 KO 号可能会涉及到多个通路。
KO 号不代表通路,因此这里有一种可行的办法就是把 KO 号给映射到 KEGG 通路上,也就是在 KO 注释的基础上进一步给它分配 KEGG pathway 的注释。
但一个 KO 号代表的是一类基因,可能会出现在多个通路里,而这多个通路可能并不会在同一个物种中出现,比如说一个 KO 表示的基因它可能在植物中参与到了一个植物独有的代谢通路中,但是在动物中它就没有这样的通路。
因此在这里我们需要选择专门的物种进行注释,注释的方法就是从 KEGG 的官网上下载各个 pathway 与 KO id 的对映文件。
首先 KEGG 数据库所涵盖的物种列表在 https://www.genome.jp/kegg/catalog/org_list.html 中。
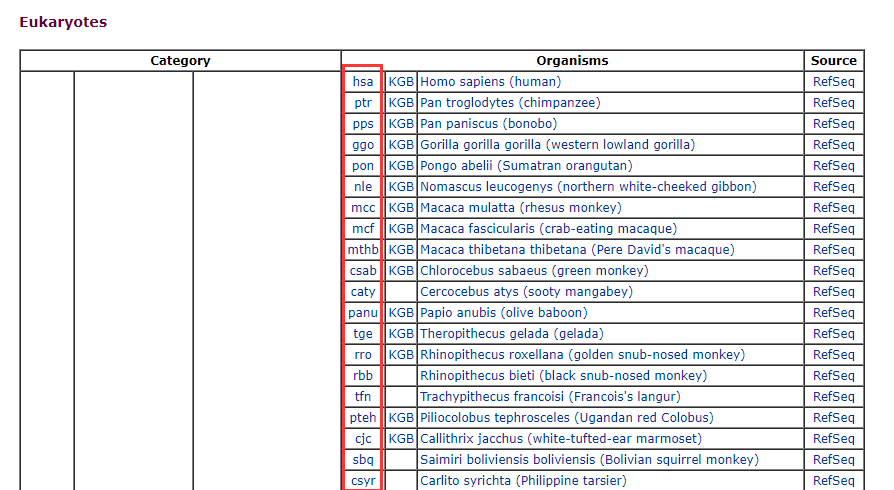
红框所示的部位为各个物种对应的物种编号,然后可以输入下列网址进行下载:
1 | http://www.kegg.jp/kegg-bin/download_htext?htext=物种编号00001.keg&format=htext |
比方说我要下载人类的映射关系文件就可以输入:
http://www.kegg.jp/kegg-bin/download_htext?htext=hsa00001.keg&format=htext
总库(拥有所有物种的所有映射关系)的下载:
http://www.kegg.jp/kegg-bin/download_htext?htext=ko00001.keg&format=htext
下载以后的文件是长这样的:
1 | A09100 Metabolism |
这里的等级层次类似于 GO,以 A 为开头的行代表 KEGG pathway 的 “大类” ,以 B 为开头的行代表二级分类,以 C 为开头的行则代表着不同的通路,后面的名字对应通路名称,其下以 D 为开头的行代表可以注释到这个通路的所有基因和对应 KO 号(一个 KO 号可能对应多个通路)。
因此使用 Kofamscan 得到 KO 注释文件后,可以使用对应物种的 pathway-KO 文件给注释相应的 pathway,根据注释信息进行后续富集分析。如果在 KEGG 的官网找不到目标物种,也可以选择一个近缘的进行分析,同样可以提高不少准确性。
代码在上面提到的知乎教程中那个老哥写的非常好,如果友们有需要也可以去下载一份然后根据个人需求修改。
后记
更新(来自 2023.1.11):今天把 KEGG 注释的部分也给补充上了,应该算是补充上了一个相当重要的部分,后续看情况找时间补充 Pfam、nr 和 CAzy 注释的内容,但这几种注释相对于 GO 注释和 KEGG 注释而言会更简单方便,有了 GO 和 KEGG 的经验以后自行学习其他的注释理应上来说是不会存在太大问题的。
更新(来自 2023.7.03):精简化了很多无用的内容,真想给喜欢说废话的自己来上一拳。
更新(来自 2024.8.24):完善了一些信息,避免可能导致的混淆。