比较转录组分析(三)—— 组装的质量检测与去冗余
前言
前文提到怎么组装数据,这里探讨下如何判断组装质量。
讲完组装质量后,再讲一讲去冗余的概念以及为何要去冗余,去冗余的方法有哪些等。
更新日志
2022.11.22 补充了在进行后续分析前给 Contig 添加物种名前缀的内容,补充了后记内容,添加了部分粗体。
2022.12.06 补充了一些内容
2023.07.03 精简了内容
组装质量评估
N50 —— 评估指标
计算 N50 时,首先会把组装中所有 碱基 的个数统计出来,比如 ATCG 就是四个碱基。
统计出来后,将 Contig 按照它们的长度依次排列,从长到短。
排列完以后,从最长的 Contig 开始,计算它的碱基数量并加和起来,当计算到某一条 Contig 时所加和起来的碱基数量到达了组装所有碱基数量的一半时,这一条 Contig 的长度就是 N50 的数值大小。
N50 可以反应组装的细碎程度,当其很小时,表明组装出来的 Contig 整体较短的。
想要统计这一系列组装信息可以使用 Trinity 自带的脚本进行(低版本没有):
1 | locate TrinityStats.pl # 如果你不知道位置可以使用这个。 |
Bowtie2 比对率
准确来说,Bowtie2 的比对率反应的是这个组装对于测序数据的代表水平,它的比对率越高,表明 Trintiy 使用测序数据组装的越完好,这个可以作为一个参考。
比对的方法请见网上教程,在后续事实上也会用到 Bowtie2 去比对定量,因此这一步可以放在后续的流程中顺便完成。
比对出来的信息是长这样的:
1 | 27044147 reads; of these: |
该例中取消了不和谐比对的统计(默认参数是会将不和谐比对纳入总比对率中的)。
快速得到总体比对率:
1 | cat bowtie.out | grep "overall alignment rate" |
BUSCO —— 应该算是金标准
BUSCO 相较于上述两者会直观很多,它的全称叫做 Benchmarking Universal Single-Copy Orthologs 。
原理:它有不同类群物种的保守基因数据库,通过比对判断组装的转录本里包含了多少这些基因,通过这一指标去评判组装的完整性。
这个软件教程非常成熟,比如:BUSCO - 组装质量评估 from 简书 或者 知乎 。
BUSCO 自带画图脚本,跑完后的结果就是长这样的(以上面教程中的为例):
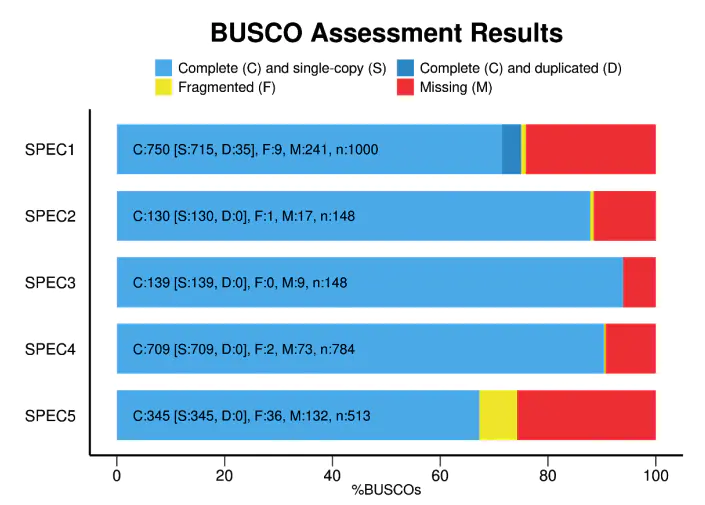
实际结果中可能深蓝色的部分(多拷贝)占比会很多,但不用担心,因为转录组存在冗余是很正常的。
BUSCO 的结果是判断一个转录组是否能作为物种参考转录组的一个很好的参考,如果在 BUSCO 结果中大多数的基因都未被找到,那么基于这个转录组的分析也会变得不是那么可靠:
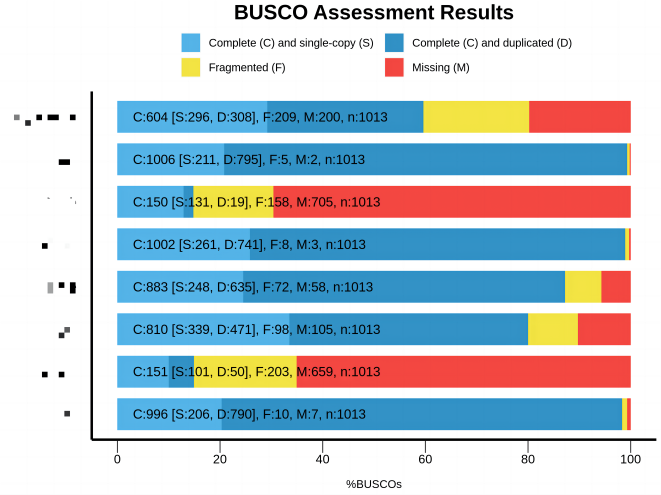
具体的代码执行:
1 | busco --offline --lineage /path/arthropoda_odb10 -i xxx.fasta \ |
--offline 和 --lineage 是直接让 BUSCO 离线工作,基于本地的数据库执行命令。
数据库自己下载会方便很多,虽然 BUSCO 可以指定谱系然后在线下载对应数据库进行比对但是远没有自己下载来的迅速。
BUSCO 不同 lineage 的下载地址:这里。
BUSCO 值得注意的点
- BUSCO 有不同的数据库,要根据自己的需要下载,如果找不到自己要分析的物种类群的话也可以使用真核生物数据库进行。
- BUSCO 的绘图脚本虽然方便,但它的宽 & 高是确定的,可以自己更改脚本进行对应修改,同时也可将输出文件改成 pdf 格式,该脚本基于 R 进行绘制的,R 输出的 pdf 为矢量图,十分清晰。此外 pdf 也便于编辑和修改,后期润色处理会更方便。
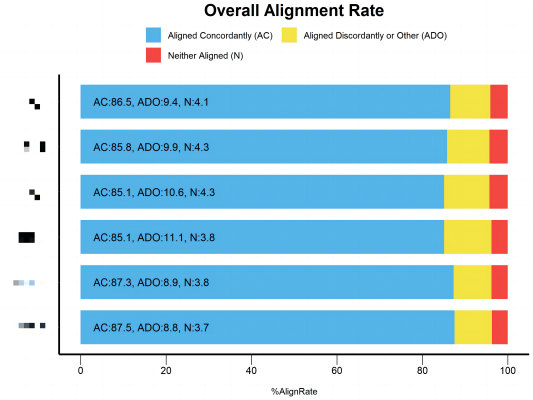
- BUSCO 的脚本画的图还挺好看的,如果别的数据也类似于这种格式,可以考虑把这个脚本改成需要的样子,例如我用这个脚本画 Bowtie2 的图:
其他评估组装质量的方法
除了上述提到的方法以外,很多指标也能够当作评估的参考,比如全长转录本的含量、ExN50等等。
甚至有对应的软件可以给组装打分,比如说 TransRate ,原理其实就是总结了上述几个方法然后根据上述几个方法的结果进行综合评价。
组装的前缀添加
如果在组装后仅使用这个组装进行后续的分析,前缀就没有太大的必要添加。
但如果后续需要利用转录组进行系统发育分析的话,添加前缀的必要性是很高的,因为这涉及到后续一系列序列的处理和串联,如果一开始没有加上那么后面会有很多麻烦,而且加上了也会让转录组组装具有物种标识,在之后的分析中不容易弄混。
对于刚跑出来的组装就可以进行这一个处理了,具体的代码如下:
1 | import argparse |
这是我挺久以前写的一个脚本,因为网上的脚本大多只涵盖到了一个功能(比如说只能简化 id 或者只能加前缀),所以我写了个可以加前缀、简化 id、加后缀于一身的脚本。下载地址请见我的 github 或点击此处。
具体的使用例(加前缀):
1 | python id_modification.py -m pre --string Human@ -f before.fasta -o after.fasta |
--string 后的字符串一定要以 @ 结尾并且有且仅有一个 @ (当然使用下划线 _ 也行,但所有物种应当统一使用其中某一个)。
处理后的 fasta 文件前面就都有 Human@ 了,这样做的具体用处在之后的分析中会一一提到。
组装的去冗余
①、为什么说组装有冗余:
正如之前所说,无参转录组的组装中,很大概率多条 contig 是同属于一个基因的,而且就算这些 contig 是所谓预测出的可变剪切 isoform,它也并不真实可靠。
②、有没有必要去冗余:
Trinity 的官网上是这么说的:
- Lots of transcripts is the rule rather than the exception.
- Most of the transcripts are very lowly expressed, and the deeper you sequence and the more complex your genome, the larger the number of lowly expressed transcripts you will be able to assemble. Biological relevance of the lowly expressed transcripts could be questionable - some are bound to be very relevant.
- There's really no good reason to immediately filter them out. They can be 'passengers' throughout all of your data analyses, and if any of them are important, they'll ideally surface in the relevant study. You can put them all through Trinotate.github.io for annotation/analysis, and you can put them through DE studies just fine (those with insufficent reads will get directly filtered out during the DE analysis protocols to avoid problems associated with multiple hypothesis testing); If the read counts are few or lacking, they simply won't surface as significant DE entries, but if there's protein homology or other interesting features, you'll want to continue to capture this info - hence don't feel the need to immediately filter!
简单翻译一下就是,有很多转录本是很正常的,没有充分的理由去把它们马上过滤掉,因为可能它们中的某一些是很重要很有价值的。
当然,绝大多数人是会进行去冗余的,因为这样做会有这么些好处:减少后续分析所需要的内存资源、提高 reads 映射的准确性、所留下的转录本更具有代表性等等。
方法① —— 取最长转录本
顾名思义,将 Trinity 组装出来的同属于一个基因的不同 isoform 中挑取最长的出来作为代表转录本。
实现方法(基于 python):
1 | import sys |
下载地址在这里,对应用法如下:
1 | python longest_contig.py xxx.fasta longest.fasta |
脚本思路比较简单且仅适用于 Trinity 的输出结果,想在 windows 系统上进行可以考虑使用我弄的 JuseKit(??)。
方法② —— 对组装进行聚类去冗余
这里涉及到了一个由国人大佬开发的软件叫 CDHIT,原理和教程请移步 CDHIT 聚类算法 from 简书。
我使用的代码(参考了 Trinity 官网推荐的严格聚类):
1 | cd-hit-est -o cdhit.fasta -c 0.95 -i xxx.fasta -p 1 -d 0 -T 5 -M 10000 -b 3 |
-c 为相似度阈值,越高时越严格。
-b 为对齐带宽,越低时越严格。
事实上不加 -b 用默认值也是可以的,但是我一开始就用上了于是乎后面就一直在用。
去冗余后我们就会把组装认为是由非冗余基因组成的 Unigene 库并用于后续的分析(当然完全不冗余是不太现实的)。
以前生信课里跑数据的时候,我非常纠结去冗余的方式到底该选哪个好,所以我问了老师,他的回答如下:
有些人去冗余他会采用非常极端的方法,就是把所有的去冗余方式都走一遍,但是这也并不一定算是严格正确的。生信的流程并没有一个统一的标准,你觉得哪个好那就选择哪一个,因为哪种都有它的优点和缺点。
就比如说如果你不去冗余,可能一个蛋白编码基因它的一个 isoform 是上调的,另一个 isoform 是下调的,总的来看的话这个基因就是没有变化的,但是又有说法说更长的 isoform 可能代表了更高的活性,这一系列因素软件是不可能完全考虑到的。去冗余了,也会有之前提到的那几个小问题。
从这些地方就可以看出来,每个步骤里包含的学问都很多。但不知道怎么办的话,流程千千万,选一条适合自己的就足够。
后记
关于流程选择的问题,我个人感觉如果课题组里已有师兄师姐做过相同的分析并且发表了文章,那么就跟着课题组前辈们的流程走就好。如果没有的话,那么选一篇自己喜欢的论文照着它的流程走也不失为一个很好的选择。
如果有时间的话...... —— Juse