比较转录组分析(二)—— 原始数据的质控与转录组组装
前言
这次文涉及到的主要是原始数据的质控与转录组组装,包括一些小知识例如如何判断数据受污染。
更新日志
2022-11-18 补充了在 for loop 和 while loop 中使用 & 的需警惕之处。
2022-11-21 补充修改了部分内容。
2022-12-05 更新了物种参考转录组的一些小知识。
2023-07-03 精简了内容。
原始数据的质控
提醒
后续所提及的测序数据皆为二代测序的双端数据。
二代测序的原理可以搜索 illumina 的下一代测序(NGS)原理介绍视频观看学习(illumina 的测序特点在于桥式扩增,其他测序公司可能略有不同但原理上是一致的,例如 BGI 的滚环扩增)。
所需要的软件有 FastQC、MultiQC、Trimmomatic、Cutadapter(可选)。
需要提前了解的:
- 测序数据文件中每一行所代表的意思。
1 | @V350095853L2C001R0010000000/1 |
以这一段数据为例,第一行 @ 后续的字段为序列名称,通常在双端数据的两个文件里一个以 /1 结尾,一个以 /2 结尾。其后的三行是碱基序列及其对应的碱基测序质量(有两种质量得分模式,大多数采用 Phred-33)。
FastQC & MultiQC
作为测序数据质量评估的老牌软件,fastqc 的网上教程已经非常丰富及完善,这里我着重提其中最需要注意的部分。
1 | fastqc -t 6 -o out_path sample1_1.fq sample1_2.fq |
在这里我给出一个并行命令以节省时间的技巧:
比如当所有双端数据文件都以 _1.fq 和 _2.fq 结尾时,假设我的目录下有(此处以压缩格式为例):
1 | ls ./ |
那么我可以这么做以并行命令:
1 | for i in A B C; |
注意!如果在 for loop 或 while loop 中所需要运行的命令量非常惊人时,不可使用 &,否则可能造成服务器宕机。
运行后,fastqc 会输出一份有关测序数据的质检报告(html 格式,每个测序文件一个),里面最需关注的可能先是序列的碱基质量,其次是其中是否含有接头序列,其他的报告应当分情况看。
例子(以上述知乎教程中为例):
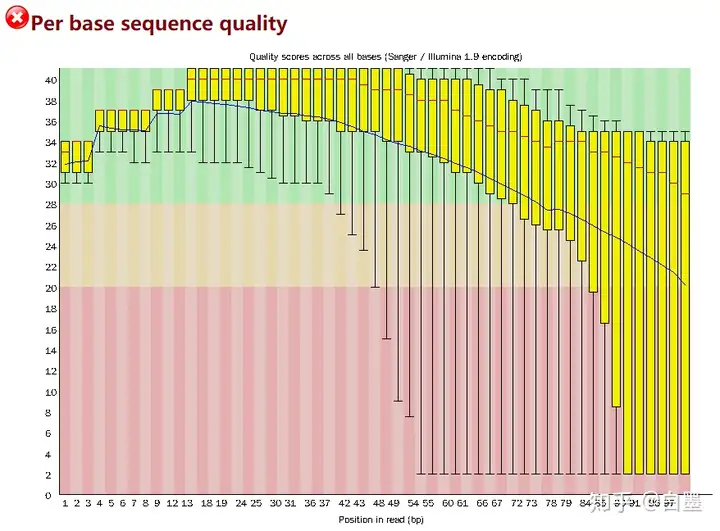
让我们来着重讲一下其他可能需要关注的地方。
GC 含量
GC 含量能反应什么?事实上对于一种特定的物种而言,它的基因组整体的 GC 含量是相对稳定的,因此所有的序列其 GC 含量会呈现一个正态分布的峰状。因此当这个图存在异常时可能说明了这么几点。
例子(正常的峰):
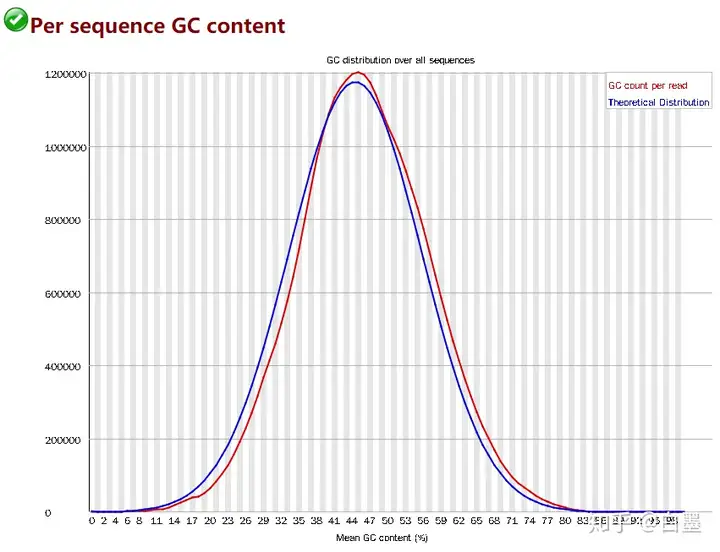
Contaminated library(文库受污染)
当除了一个很大的峰以外还存在尖峰,说明文库可能混进了接头二聚体(可能性不高)。
当出现一个宽峰,说明文库可能混进了其他物种的序列。
Biased subset(序列偏好)
当除了一个很大的峰以外还存在尖峰,可能代表了文库中某类序列较多,这对于转录组来说是比较正常的,因为有部分 mRNA 的表达量可能会非常高。
此外很多因素也会产生一定的偏差,例如测序时的偏好、PCR 的扩增偏好、在建库过程中利用 PolyA 富集真核生物 mRNA 时可能由于 RNA 的降解导致大部分得到的序列都靠近 3 '端等等,但这些应该都不会显著影响 GC 含量的整体正态分布。
重点
一般而言,转录组的数据在 fastqc 这里确定有没有污染是不够准确的,我们可以把它当作一个警示,但有些时候,正如上述所说,它出现的警告可能只是转录组会出现的正常现象之一。
或许有用的小知识
要确定有无其他物种污染,我们可以看后续使用 Trinity 组装出来的转录组有多大,如果过大的话则说明组装出了多个物种的 contig,要更加精确的判断可以通过 diamond against nr database,提取出 contig 的 taxonomy id 信息,如果大量存在另一类物种的 contig,可以在 NCBI 上下载该物种的基因组并使用 STAR 比对原始数据到基因组上确定 Uniquely mapped reads 和 mismatch rate。
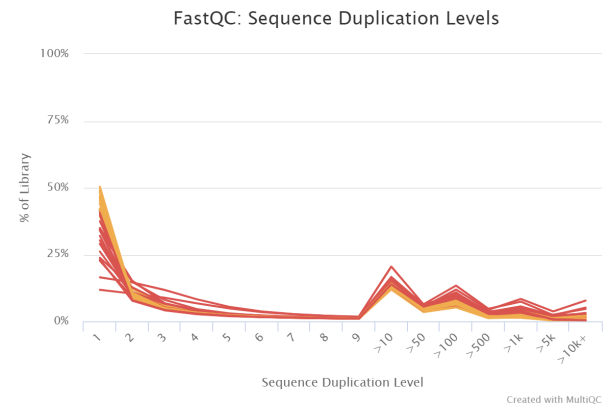
序列重复等级
转录组测序的数据在此处基本都是会爆红的,所以不用太担心,毕竟某个高表达 mRNA 的序列会在这里重复上千甚至上万次。
MultiQC 执行
multiqc 用于整合所有质检报告,将所有 fastqc 跑出来的 html 放在一个文件夹后运行以下命令:
1 | multiqc . |
此后,它会将所有的报告整理输出一份整合报告,但请注意,不同类型数据的质检报告它是没法整合在一起的(如 50bp 的数据和 150bp 的数据)。
此外,multiqc 除了整合 fastqc 的结果外还可以整合许多其他的输出结果,内容见下表。
详细的教程见:整合QC质控结果的利器 —— MultiQC from 简书。
可能你会感兴趣的事
事实上 fastqc 和 multiqc 作为质检工具,虽然它常被用到,但是它的结果很少被直接放在论文之中,很多文章可能会简单地提一句经过 fastqc 和 multiqc 质检数据一切 ok 之类的话,如果要把图放进论文的话可能会要分别标明 multiqc 结果中每个线属于哪个样本之类的(亦或是放进补充材料里)。
Trimmomatic & Cutadapter —— 质控工具
多数测序数据从测序公司经手到客户手上时一般都被良好地处理过了。
例如华大它会使用自己开发的 SOAPnuke 对原始数据进行一次数据过滤。
所以我们对这些 clean data 可以选择:①、不做任何处理直接进行分析;②、再跑一次 Trimmomatic。
我的老板是这么说的:通常我会自己跑一次 trimmomatic,反正不用太多时间。
Trimmomatic 执行
简易版教程请见:生信软件 | Trimmomatic (测序数据质控) from 知乎
代码(此处使用了 Trinity 自带的 trimmomatic 默认参数,基于关于高通量 mRNA 序列数据的最佳修剪设置):
1 | nohup trimmomatic PE -phred33 input_forward.fq.gz input_reverse.fq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:5 MINLEN:25 |
推荐加一个 nohup,这样 trimmomatic 的修剪日志会输出到 nohup.out 中,你可以在其中查看 reads 的修剪情况,而这个情况一般是可以在 paper 里提一嘴的。
例:
1 | Input Reads: 9692546 Surviving: 9123724 (94.13%) Dropped: 568822 (5.87%) |
需要注意的是,Trimmomatic 只能识别和去除 Illumina 测序数据中的接头。
如果你的数据是在其他测序公司里测得的并且未经过处理,那么你或许需要使用其他的软件(例如 TrimGalore 或者 Cutadapter)。
转录组的组装 —— Trinity
作为一个在不断更新老牌 de-novo assemble 软件,目前 Trinity 在无参转录组的组装方面已经有了相当强大的性能,其附带的完整预包装 pipeline 也使得仅使用这一个软件便可以完成绝大多数的无参转录组分析(仅需配置好其他所需软件即可)。
其他的软件如 SPAdes、TransABySS、SOAPdenovoTrans 大家有兴趣也可进行了解。
Trinity 执行
基础教程在网上非常多,最详细的引路:Trinity Github wiki
组装原理简单来讲就是根据序列的 overlap 关系将其拼接成 contig。
命令:
1 | nohup Trinity --seqType fq --SS_lib_type RF --max_memory 40G --trimmomatic \ |
tip:Trinity 能够使用参数 --trimmomatic 调用软件进行原始数据的过滤,但是建议自己跑 Trimmomatic 以后再写对应的 sample file,不要在此处调用。不过如果仅需组装转录组,那么可以忽视这一条建议。
--SS_lib_type
需要格外关注的是 --SS_lib_type 参数,当测序数据文库是通过链特异性建库得到的,那么这里就需要指明链特异性。
不知道测序数据是否为链特异性:检查 reads 链特异性 from Trinity
了解链特异性建库:说一说转录组链特异性文库的那些事。(定量更准确,可看下图)
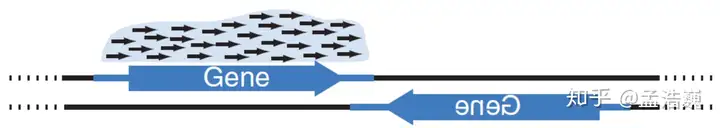
链特异性文库如何构建:RNA 的第一条 cDNA 合成后,在第二条 cDNA 合成时用 dUTP 替换 dTTP,因此第二条链中的 T 都变成了 U,接上接头后,使用酶特异性降解带 U 的链,因此只保留了第一条链,并具有明确的方向。
大部分数据都是非链特异性的,但如果不知道具体情况最好进行检查。
--samples_file
如果后续需要进行差异表达分析,那么一开始组装时便使用这个参数会大大节约功夫,因为它是后续分析中 Trinity 所要求的必需输入文件(此处为可选)。
文件内容示例:
1 | cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq |
其中第一列代表不同的处理,第二列代表处理的不同生物学重复,后两列为双端数据的位置(制表符分列)。
在使用该文件后,Trinity 会自动将所有双端数据合并进行转录组组装。
其他参数的介绍可以移步 github 官网或 使用Trinity进行转录组组装 from 简书。
Trinity 的输出
1 | >TRINITY_DN417671_c0_g1_i1 len=249 path=[0:0-248] |
以 > 开头的为 Trinity 组装出来的转录本,我们也可以称它为 contig。
以上述中的 TRINITY_DN417671_c0_g1_i1 为例。
里面的 TRINITY_DN417671_c0_g1为基因编号,而其后接着的 _i1 则是 isoform 的含义。
一个基因可以有不同的剪切方式,这种可变剪切会使得一个基因能产生多个转录产物,而 Trinity 则根据测序数据对基因的可变剪切进行了 “预测”。
这里的预测打双引号的原因在于 Trinity 的 isoform 并不完全精准,因为仅凭转录数据而脱离基因组的无参组装里,并不可能准确的判断出可变剪切情况,因此可变剪切分析一般都要求参考基因组。
一些题外话
有些时候,用于差异表达分析的数据也可以用来组装转录组,有些时候则不行。
因为如果要得到一个物种的参考转录组,一般来说会从它身体的多个组织中进行 RNA 提取和等摩尔混合测序,并且使用更高的测序深度,原因如下:
①、不同组织的表达景观不同,有些 mRNA 在某些组织中可能含量非常少,而在另外一些组织中则含量很多。所以取多个组织提取 RNA 测序可以保证获得信息足够全面的转录组数据。
②、更精的测序深度有利于稀有 mRNA 的识别。
但是用来做差异表达分析的测序数据往往是从某个特定的组织中提取的。所以可以看到一些研究中,研究者们采取 参考转录组数据 和 差异表达分析数据 分开来的策略。例如,用来组装物种参考转录组的数据是双端 100bp 的,但用于差异表达分析的数据则是单端 50bp 的。
注意:参考转录组 ≠ 有参转录组。
参考转录组指可以代表一个物种整体转录 mRNA 情况的组装。
有参转录组指具有参考基因组的物种的转录组组装。